










In 1812, John Warren published a description of chest symptoms that he called angina pectoris, without knowledge of the underlying pathogenesis, although, at the time, coronary ‘ossifications’ were being noted during anatomical dissections.1 In the late 19th century, physiologists noticed that occlusion of the coronary artery of a dog resulted in ‘quivering’ of the ventricles and was rapidly fatal.2,3 Around this time it was suggested that coronary thrombosis was the cause of MI.4 In the early 20th century, ECGs showing ST segment change were being used to help diagnose MI.1 By the mid-20th century, the introduction of coronary angiography allowed the natural history of coronary artery disease (CAD) and acute coronary occlusions to be observed.
Aspartate transaminase was the first cardiac biomarker to be used in clinical practice in the 1950s, and was one of three criteria, along with ECG changes and symptoms, in the 1959 WHO definition of MI.5 In the late 1970s, the wave front phenomenon of myocardial necrosis over several hours after coronary artery occlusion was observed in dogs, and around the same time the pathophysiologic mechanism of plaque rupture/erosion triggering thrombotic occlusion was being developed. Since the recognition of the pathophysiological mechanism and the development of targeted reperfusion therapies, mortality in acute ST-elevation MI (STEMI) has reduced from 18% (control group of the GISSI-1 trial in 1986) to 4% in 2006.6,7
In 1979 the WHO added creatinine kinase (CK) as a recommended biomarker for diagnosing MI, followed by the specific CK myocardial band (CK-MB) isoenzyme, which is much more prevalent in cardiac than in skeletal muscle. The development of immunoassays in the 1980s enabled measurement of CK-MB mass, which allowed earlier detection of myocardial damage, although specificity remained an issue. Attention turned to the contractile apparatus of cardiomyocytes, and after disappointing results with myosin light chains, cardiac troponin (cTn) was first discovered in 1965 by Ebashi and Kodama.8 Katus et al. demonstrated its specificity for myocardial cell damage in comparison with CK-MB in 1991.9
In a similar pattern to the current situation with high-sensitivity troponin (hsTn) assays, large numbers of studies in the 1990s showed that significant numbers of patients classified as having ‘unstable angina’ by WHO criteria actually had elevated cTn.5 The changed definition of MI to include cTn as the preferred biomarker in 2000 was met with concern initially regarding the increase in the positive rate. However, this was replaced with widespread acceptance as biochemical parameters to reduce assay variability were introduced, leading to endorsement of cTn as the biomarker of choice in the first universal definition of MI (UDMI) in 2007.10 The UDMI also introduced the concept of type 2 MI, and the fourth UDMI in 2018 further developed the concept of myocardial injury, with the recognition that myocardial damage as indicated by the new hsTn assays could frequently occur without ischaemia.
Type 2 MI
MI by definition refers to necrosis of cardiomyocytes due to ischaemia.4 Type 2 MI refers to those cases in which this is due to an imbalance between supply and demand, in contrast to that due to an acute atherothrombotic event.11 Being ultimately a pathophysiological distinction, this continues to create difficulty in the definition of different types of MI in clinical practice, given the resulting heterogeneity in the literature. Systemic conditions such as sepsis can also be associated with more type 1 MIs (acute plaque events) than type 2 MIs, and this has important prognostic and treatment implications.12–24
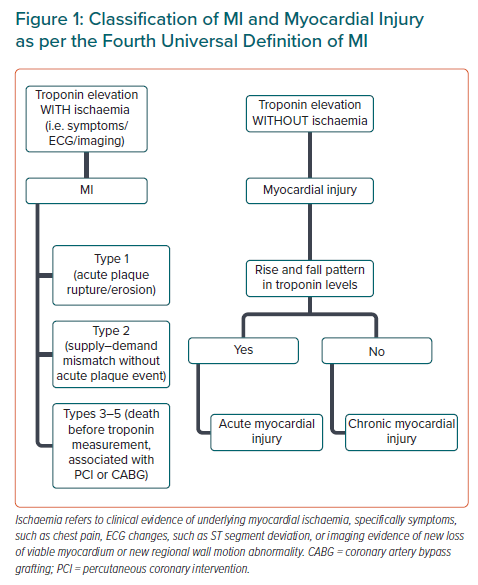
Type 2 MI occurs more frequently than type 1 MI.25 Type 2 MI is common in hospitalised patients, on average accounting for 10–20% of MIs.26 Its causes are myriad and range from acute cardiac conditions such as a tachyarrhythmia to non-cardiac conditions, such as anaemia. Complex molecular and cellular signalling pathways are triggered once the cardiomyocyte is exposed to ischaemia, and results in cell death mainly via apoptosis and necrosis. These processes ultimately result in the presence of troponin in plasma, the cornerstone of the UDMI.
Current Definitions
Troponin was incorporated into the first definition of MI by the European Society of Cardiology (ESC) and American College of Cardiology (ACC) in 2000. A rise and/or fall in troponin is required for the clinical diagnosis of all types of MI, along with any one of the following features of clinical ischaemia: symptoms (no duration defined in fourth UDMI), ECG changes, or imaging evidence of new loss of viable myocardium or new regional wall motion abnormality.27 For the first time in 2007 subtypes were introduced, including type 2 MI.
In the most recent fourth UDMI, the concept of ‘myocardial injury’ is further developed as separate from type 2 MI in that there is an absence of evidence of clinical myocardial ischaemia despite an elevation of troponin.11,28,29 If there is an appropriate rise and/or fall in troponin, the myocardial injury is considered acute (Figure 1); and chronic if the levels are stable (<20% change). In type 2 MI, as in all types of MI, there must additionally be evidence of clinical ischaemia. Troponin elevations alone, regardless of other clinical features, are prognostic of both cardiac and non-cardiac outcomes.30,31 The presence and magnitude of clinical ischaemia is affected by many factors such as the severity and nature of the concurrent illness, comorbidities, and the degree of underlying CAD.
One of the most common differences in the literature with regards to the definition of type 2 MI is clinical evidence of ischaemia and whether sepsis is present (sepsis is excluded in the UDMI as a cause of type 2 MI). Due to heterogeneity, it is difficult to quantify the incidence of type 2 MI as a proportion of all MIs.26,28,32–36 For example, one study using the UDMI and focusing on only coronary care unit/intensive care unit patients reported a 7% incidence, while another study in emergency department patients presenting with elevated troponin reported a 35% incidence.4,28,37 A large study involving almost 5,700 hospitalised patients showed that 62% had an abnormal hsTn, and there was dynamic change in 24%. However, only 6.1% had a final diagnosis of type 1 MI, suggesting that up to 17.9% may have had a type 2 MI or acute myocardial injury.38
More recently, the term MI with non-obstructive coronary arteries (MINOCA) has been used in the literature, including in an ESC position paper, referring to lesions with <50% stenosis.30,39–42 A US consensus statement has considered having an additional functional assessment that is, fractional flow reserve (FFR) >0.80 as a criterion. Crucially, this diagnosis can only be made after confirmation of the diagnosis of MI and the performance of coronary angiography. It is an exclusion diagnosis, encompassing many conditions and including both type 1 and 2 MI. Myocardial injury, myocarditis and takotsubo syndrome do not come under the terminology because they are not MI.
Large MI registries show an incidence of MINOCA of 6–13%.41 Type 1 and type 2 MIs are separate but overlap with MINOCA, in that both can occur within and outside the MINOCA definition. In a recent review, type 2 MI comprised 10.5% of MINOCA.43 The unique feature of this term is that knowledge of the coronary anatomy is required, and therefore it usually captures a cohort that has been referred for invasive coronary angiography and who are generally healthier than those not referred for angiography. The COVID-19 pandemic has seen an increase in the number of MI presentations, including STEMI, with up to 40% with normal coronary arteries.44 Further intravascular ultrasound (IVUS) and optical coherence tomography (OCT) imaging studies are required to define the pathophysiology.
Aetiology of Type 2 MI and Myocardial Injury
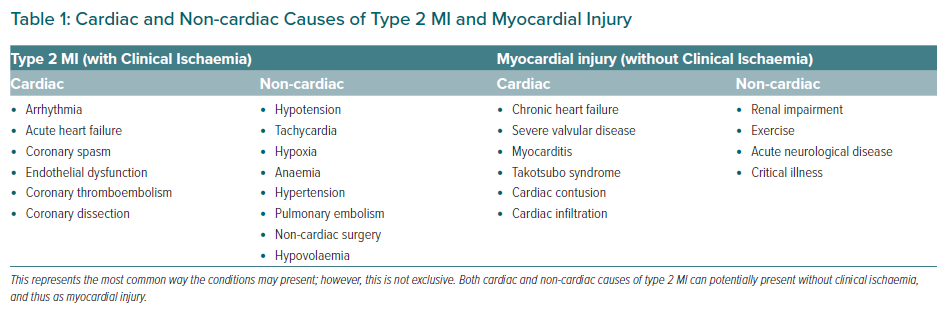
There are a myriad of cardiac and non-cardiac conditions that can upset the balance between oxygen supply and demand in the myocardium and cause a type 2 MI.45 Table 1 lists these, along with the causes of myocardial injury, which by definition are non-ischaemic. There are a few common denominators of type 2 MI that can be a result of many of the listed clinical conditions: tachycardia, hypotension, and hypoxia. We postulate that these may be part of the final molecular mechanism by which type 2 MIs occur.
MI after non-cardiac surgery is a unique clinical scenario in which many potential mechanisms may contribute to both type 1 and type 2 MIs. Bleeding, hypotension, hypoxia, hypothermia, tachycardia, micro-embolism in the coronary circulation, catecholamine surges, and diastolic dysfunction due to preload alterations causing subendocardial ischaemia, may all contribute to the occurrence of type 2 MI perioperatively.32,34,46 These may or may not occur in the setting of pre-existing CAD of varying severity, the presence of which usually portends a worse outcome.47 Although both type 1 and type 2 MI are possible on a pathophysiological level, it is thought that the majority are type 2.
Heart failure is another interesting entity that may be associated with different mechanisms of troponin release.48 A non-dynamic pattern in a stable patient may be related to chronic myocardial injury that is non-ischaemic, whereas an acute rise may be due to type 1 or type 2 MI. Type 2 MI in heart failure may be mediated by small vessel CAD, increased transmural pressure with increase in left ventricular end-diastolic pressure, endothelial dysfunction, or subendocardial ischaemia.34
Pathological Mechanisms
Myocardial necrosis in type 2 MI occurs from either increased myocardial oxygen demand or decreased supply, or both. Supply is determined by the oxygen-carrying capacity of blood and coronary blood flow, while demand is largely determined by systolic wall tension, contractility, and heart rate (Figure 2). The presence of CAD may play a role, altering the threshold for myocardial ischaemia in any given patient. It has become clear in recent times that plaque growth to the moderate–severe range is the result of one or more subclinical rupture events with efficient lysis and healing, such that patients with type 2 MI and significant stable CAD may actually have had silent plaque rupture events in the past.49 Individual differences in the ability to maintain coronary perfusion under stressful conditions such as critical illness also plays a role.
At the cellular level, it is probable that cardiomyocytes respond similarly to supply–demand ischaemia (i.e. type 2 MI) as in acute coronary thrombosis (i.e. type 1 MI), with membrane permeability changes, release of cytosolic vacuoles, and release of proteolytic degradation products contributing to cell death. The volume of involved cardiomyocytes is localised in type 1 MI to the territory supplied distal to the plaque event, whereas in type 2 MI we hypothesise that it may be a more global ischaemic phenomenon, with some regional myocardial dysfunction depending, among other things, on the severity and distribution of coexistent CAD. This has important implications for treatment strategies.
Appropriately, in type 1 MI the focus has been on the acute plaque event in the epicardial vessel, with successful therapies now in use such as percutaneous coronary intervention (PCI), thrombolysis and anticoagulation. Therapeutic interventions for type 2 MI need to focus on the underlying aetiology, for example anaemia, hypoxaemia and arrhythmia. In type 2 MI, absolute biomarker peaks are usually lower than in type 1 MI.33,50 There are probably several variables at play, but the degree of ischaemia is likely to be lower when compared with the absolute ischaemia that occurs with complete thrombotic occlusion of an epicardial coronary artery. Relief of a total coronary artery occlusion either spontaneously or following PCI or thrombolytic therapy with troponin washout may result in higher peak troponin levels than in patients with type 2 MI.
In myocardial injury that is by definition non-ischaemic, troponin elevation may be mediated by direct toxicity from circulating cytokines, catecholamines, or vasopressors. These factors may also play a part in the aetiology of type 2 MI.33 Tachycardia is one of the ‘final common mechanisms’, and it has been hypothesised that increased heart rate may cause troponin release due to increased wall tension and stretch, from a direct mechanical stimulation of stretch-responsive integrins.9,51 This mechanism probably also plays a role in the troponin elevations seen in patients with severe hypertension and valvular disease. Direct involvement of the inflammatory process in the myocardium, such as in myocarditis, is another mechanism separate from ischaemia that leads to myocardial injury.
Takotsubo syndrome is interesting to consider in relation to type 2 MI: patients can present with all features of an MI, with ischaemic symptoms, ECG changes, rise and/or fall in troponin, and regional wall motion abnormalities on imaging (importantly not isolated to a single vascular territory). There is no acute coronary or plaque event, hence these are not classified as type 1 MIs. The full pathophysiological mechanism is yet to be understood; however, there is an abundance of evidence that sympathetic stimulation is key, and that acute microvascular dysfunction as a result plays a central role.52 Given a distinct pathophysiological mechanism related to catecholamine excess and the lack of need for a triggering medical illness, such as in type 2 MI (e.g. anaemia, tachyarrhythmia), we consider takotsubo syndrome to be a separate entity to type 2 MI. The left ventricular dysfunction can persist for months; however, it usually resolves due to activation of myocardial cellular survival pathways in the face of the catecholamine surge.
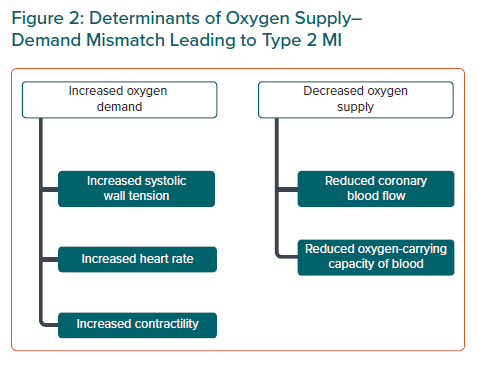
Cardiomyocyte death occurs via two major processes, apoptosis and necrosis (Figure 3).53 Traditionally, apoptosis was thought to be controlled and regulated, whereas necrosis was almost accidental due to physical or chemical stimuli.54,55 However, the discovery of molecules that inhibit receptor-interacting protein kinases (RIP1, RIP3) demonstrated that necrosis, particularly in response to ischaemia, was signal regulated (termed ‘necroptosis’). In myocardial ischaemia, although apoptosis plays a role very early, necrosis is the dominant influence, and the release of cellular contents promotes inflammation and further cell death.54
The complex molecular signalling that occurs following an ischaemic insult provides a rich source of potential therapeutic targets that may be applicable to all types of MI and myocardial injury. A few small molecules have been developed that have shown inhibition of necrosis in non-human controlled environments. Nec-1 molecules inhibit RIP1 and markedly reduce infarct size.54,56–58 Necrosulfonamide inhibits mixed-lineage kinase domain-like protein (MLKL), thus preventing the deleterious membrane effects leading to cell death.54,58
Following MI, there is a massive accumulation of neutrophils and monocytes (enhanced by extravasation of platelets and endothelial cell leakage), and a subsequent increase in fibroblasts.53,59 The extracellular matrix (ECM) of the myocardium plays an important role in the response to ischaemia. Local fibroblasts can induce further inflammation through interleukin-1, and matrikines released from the ECM initiate pro-inflammatory actions.53,59,60 The intense inflammatory response in the first few days is followed by fibrotic healing that is largely completed by 7–14 days.59 It may be that the role and type of inflammation in type 2 MI differs from type 1 MI, with higher cytokine, leukocyte and C-reactive protein levels being reported.32 Whether this represents any fundamental difference in molecular pathophysiologic pathways between type 1 and type 2 MI, or simply reflects the systemic illness setting in which type 2 MI often occurs, is not known.
Troponin and Myocardial Damage
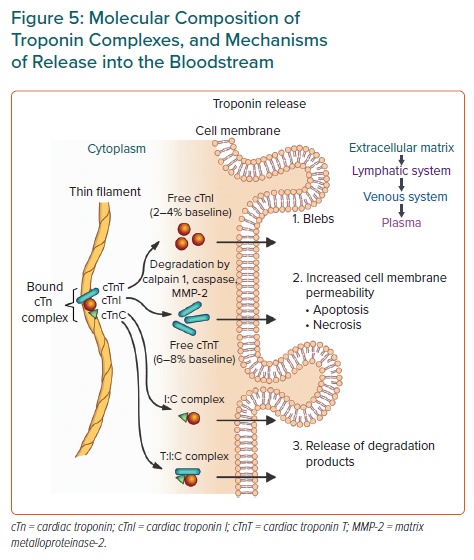
Troponin is found in all forms of striated muscle, and cTn has unique regions of amino acid sequences. This means that antibodies can be made against specific epitopes, and ultimately assays for myocardial specific troponins can be made. The cTn complex consists of three high-molecular-weight protein subunits (cTnI, cTnT and cTnC), with cTnI and cTnT the most commonly used in assays.8
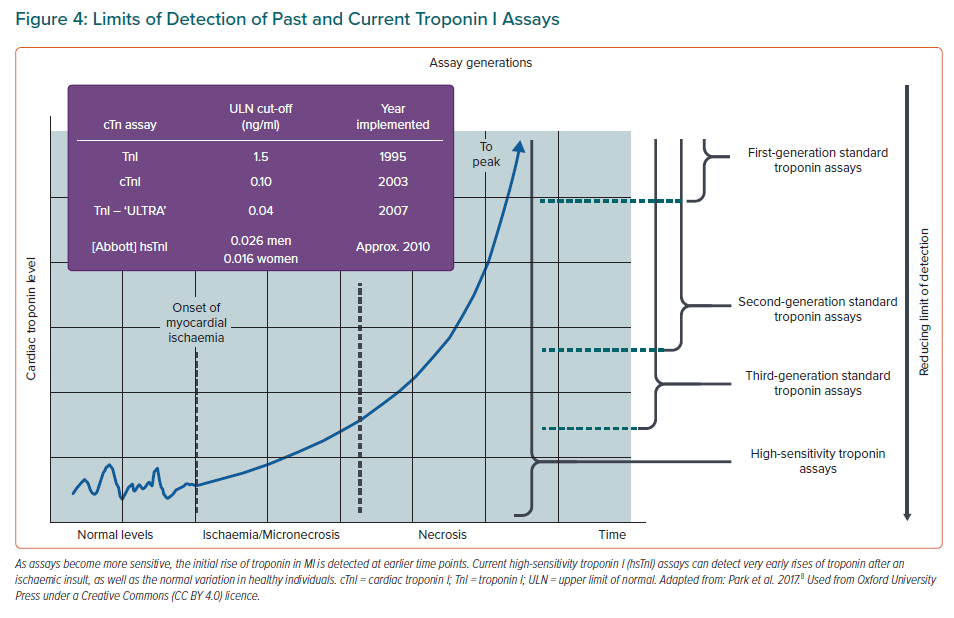
Most cTn assays are non-competitive enzyme-linked immunosorbent assays (ELISAs), using the high specificity and affinity of antibodies. After the onset of ischaemia, cardiomyocyte death can occur within 15 minutes, while histological evidence appears at 4–6 hours.54 The cTn is released from myocardium as early as 30 minutes following ischaemia. In MI, cTn peaks at the 24-hour mark, then reduces over the next 5–10 days. cTnT appears primarily as a mixture of free forms and a T:I:C complex, and cTnI appears primarily as the binary I:C complex. The first troponin assays were introduced to clinical practice in 1995, but they took 10–12 hours to become positive after an event due to the relatively high absolute minimum concentration of troponin that was able to be detected (i.e. they lacked sensitivity).38,61 Standard troponin assays have improved with time but the new high-sensitivity assays are able to detect troponin at a 10-fold lower concentration, allowing for earlier results. Some assays are able to detect troponin within 90 minutes of an index cardiac event (Figure 4).62
cTnT has a biphasic release profile. Release is initially from the cytosolic pool (approx. 10%) and is usually free-form, whereas the subsequent peak and sustained elevation is from the structural pool via degradation (by calpain 1, caspase or matrix metalloproteinase-2) of the contractile apparatus, and is mostly the complexed forms of troponin (Figure 5).8
Apart from apoptosis and necrosis, some other mechanisms by which troponin can be released from cardiomyocytes include normal cell turnover, release of protein degradation products, increased cell membrane permeability and membranous ‘blebs’.60 It is controversial whether troponin may be released without irreversible cell death, however, these mechanisms provide potential avenues for this.8,63 The idea of cytosolic versus structural troponin has been used to distinguish between reversible and irreversible forms of myocardial damage. Normalisation of troponin within 24 hours suggests a lack of ongoing cardiomyocyte structural degradation given that the half-life of troponin is 2–4 hours, with perhaps only cytosolic troponin having been released, and thus may represent a more reversible type of myocardial damage.51 However, some studies showing that the structural troponin pool is not as resistant to degradation and release as previously thought, and may be released early, have challenged this view.51,64
It is likely that the pool (cytosolic or structural) of troponin released, whether cell death has occurred or not, and whether the injury is reversible or irreversible, all vary depending on the particular circumstance. Acute hypertension resulting in a mild troponin rise that resolves within 24 hours may not represent cell death, and may be reversible, whereas persistent troponin elevation beyond 24 hours in an anaemic, hypotensive patient (i.e. a potential type 2 MI) probably represents cell death, albeit perhaps in a magnitude too small to be detected by imaging or other techniques (which require 1 g of confluent necrotic myocardium).65
Clinical Presentation and Diagnosis
Type 2 MI is generally straightforward to diagnose when there is evidence of clinical ischaemia and a clear triggering factor. Type 2 MI patients may be asymptomatic, might have minimal, if any, ECG changes, and will have troponin levels that are not as high as in type 1 MI.32,33,46,66–69 ST elevation is more common in type 1 MI but can occur in type 2 MI in 5% of patients.70–72 One of the main questions at the bedside is whether it could be a type 1 MI. A small proportion of patients thought to have type 2 MI turn out to have type 1 MI detected by the presence of plaque rupture and thrombus on angiography.25,73–76 However, the sensitivity of detection of thrombus is low.77,78 The treatment implications are significant, given that there are well-established therapeutic pathways for type 1 MI that have been shown to improve outcomes, including mortality. Importantly, type 2 MI carries with it a worse prognosis than type 1 MI, with a greater proportion of non-cardiac causes contributing to longer term morbidity and mortality.28,30,37,39,71,79–83
It is likely that the literature investigating type 2 MI has involved a significant proportion of patients who do not meet strict UDMI criteria, given that patients often do not have typically ischaemic symptoms, ECG changes or new imaging evidence. The most important differentiator is the presence/absence of factors that may disturb the oxygen supply–demand balance. In the absence of any of these factors, type 2 MI cannot be diagnosed. The incidence of coexistent CAD is variable, depending on the population studied.29,37,84–87 The incidence of significant obstructive CAD in type 2 MI ranges from 40% to 78%.25,73,85,88 Older populations with greater cardiovascular risk factors tend to have a higher prevalence of CAD, as well as of type 2 MI.66
Some other clinical associations of type 2 MI are female sex, multiple comorbidities, and lower peak hsTn level than in type 1 MI.26,28,33,37,66,71 One study using hsTnT reported an average level of 618 ng/l in type 1 MI patients compared with 180 ng/l in type 2 MI patients.30 A binary score that is able to be used in the emergency department to differentiate type 1 from type 2 MI, has an area under the receiver operating characteristic curve of 0.71.37 This score assigns a single point each to female sex, absence of radiating chest pain, and a baseline hsTnI <40.8 ng/l; a score of 3 resulted in a 72% probability of type 2 MI, compared with 5% for a score of 0. This differentiation based on criteria that are not essential for the diagnosis of type 2 MI is unlikely to be helpful.
The majority of patients with a clinical diagnosis of type 2 MI do not undergo invasive coronary angiography, with rates of 20–30%.25,73,85 In some selected series of type 2 MI patients who underwent coronary angiography, acute plaque/coronary features have been described in up to 60%.25,73–76 PCI rates in this population range from 25% to 80%, perhaps suggesting that most clinicians favour intervening if a significantly obstructive plaque is seen or if FFR is decreased.73,89,90 There have been no published series using intracoronary imaging, such as OCT or IVUS, specifically in the type 2 MI population to define whether plaque rupture and thrombus are present.
Prognosis
Generally, prognosis after type 2 MI is worse than after type 1 MI, probably reflecting a more comorbid population overall with current critical illness.26,28,33,34,39,41,79–81,91,92 Retrospective studies demonstrate 1-year mortality rates of approximately 25% for patients with type 2 MI, compared with 8–12% for those with type 1 MI.28,37,72,85,93 In one study with a 5-year follow up, type 2 MI mortality (62.5%) was twice as likely to be due to non-cardiovascular causes than to cardiovascular causes.4 Although the excess mortality may be due to non-cardiovascular causes, type 2 MI may predict subsequent cardiovascular outcomes including death to the same degree that type 1 MI predicts outcomes.32,37,91
A recent large study by Raphael et al. further suggests that arrhythmia and post-surgical status as triggering factors for type 2 MI carry a more favourable long-term prognosis than hypoxia, hypotension or anaemia.91 Troponin levels, including hsTn, have been shown to correlate with poor outcomes in patients with type 2 MI.30,31 Higher troponin elevations tend to correlate with vascular death, while lower elevations correlate with non-vascular death.68
In a recent retrospective review of a total of 475 patients who had an MI/myocardial injury during admission to a tertiary centre, those not meeting the UDMI of type 2 MI, but who met the myocardial injury definition, comprised 46% of the cohort and had similar in-hospital morbidity and mortality to those with type 2 MI.32 There was no difference in the types of provoking conditions that caused myocardial injury compared with type 2 MI, while those patients who met UDMI criteria tended to have more cardiovascular risk factors or known CAD.32 This highlights that the UDMI is a pathophysiological categorisation and therefore, in the clinical context, those patients with myocardial injury and without clinical ischaemia may have an equally serious condition with equally poor prognosis.
Treatment
Despite its prevalence and poor prognosis there have been no randomised trials of treatment for type 2 MI, in contrast to type 1 MI, for which improved management, particularly in shortening the door-to-therapy time and in the development of anti-thrombotic therapy, has resulted in better outcomes.28,94–96 Randomised trials are ongoing, testing β-blockers and angiotensin-converting enzyme inhibitors (ACEIs; MINOCA-BAT; NCT03686696). Patients with type 2 MI have high cardiovascular risk, and in one study were found to be twice as likely to be readmitted at 1 year with type 1 MI than those with myocardial injury.28,37,79,97
The initial management should be to reverse the triggering factors, such as arrhythmia or anaemia. The well-established evidence base for anti-platelets and anticoagulants in type 1 MI has not been shown to be of benefit in type 2 MI, and may cause harm, particularly bleeding, in an elderly cohort. Many patients having a type 2 MI may be on cardiovascular medications, such as β-blockers, anti-hypertensives and statins. Not surprisingly, given the lack of evidence for the type 1 MI treatments in type 2 MI, studies demonstrate at most a 50% prescription rate for antiplatelet therapy, statins, β-blockers and ACEIs or angiotensin II receptor blockers on discharge for type 2 MI patients.32,33,41
Given that by definition a clinical diagnosis of type 2 MI means that the clinician believes there has not been an acute atherothrombotic event, we hypothesise that dual antiplatelets and anticoagulants are not likely to be beneficial. If there is evidence of coexistent CAD, these patients would be categorised in the recent ESC lipid guidelines as being at very high risk, and as requiring statins to reduce LDL cholesterol to <1.4 mmol/l.98
The proprotein convertase subtilisin/kexin type 9 inhibitor has been shown to reduce type 2 MI after acute coronary syndrome.99 Specific therapies targeting the cardiomyocyte signalling mechanisms following ischaemia have thus far proved elusive in humans. Nonetheless, conceptually it is likely that any specific treatments for type 2 MI will come from the cellular response to ischaemia, given that it is the final common pathway that leads to injury, whatever the initial trigger may be.
One factor that is common to the many triggers for type 2 MI is tachycardia, which, as well as potentially triggering myocardial stretch mechanisms, creates an oxygen supply–demand mismatch by increasing myocardial work (demand) and reducing diastolic time and thus coronary perfusion (supply). In the POISE trial, which used metoprolol as a preventative therapy for perioperative MI in non-cardiac surgery, there was a benefit to β-blockade in preventing MI (HR 0.73; 95% CI [0.60–0.89]; p=0.0017), as defined by the universal definition at the time, which preceded hsTn and the introduction of myocardial injury.100 Unfortunately, this was offset by hypotension and ischaemic strokes (which was thought at least in part to be related to hypotension predisposing to cerebral hypoperfusion). It may also be the case that given that most morbidity and mortality following type 2 MI is from non-cardiovascular causes, prevention of a troponin rise, and thus myocardial damage, might not greatly alter overall prognosis.
The role of invasive coronary angiography with or without PCI, as well as CT coronary angiography, is not well-defined in the type 2 MI population.101 Furthermore, IVUS and OCT have been little used. Referral for angiography is low in this cohort, which often consists of elderly patients who might have renal dysfunction, cognitive impairment, bleeding and/or anaemia. Delayed functional testing is often used, although there are no long-term outcome studies to help guide selection of the optimal strategy. Trials are currently ongoing to investigate whether routine invasive coronary angiography in type 2 MI and myocardial injury improves prognosis (ANZCTR; ACTRN12618000378224). Ultimately, in any individual case, the clinician must draw on all the information at hand to decide whether the finding of CAD would change management.
Impact of High-sensitivity Troponin
Given that hsTn is approximately 10-fold more sensitive than previous standard assays, minute release of troponin will be detected more frequently. Use of hsTn is expected to result in an increase in the diagnosis of type 2 MI, with even small elevations being recognised.28,34,79 Although a ‘rise and/or fall’ is required in the UDMI, no specific numbers are currently incorporated (the generally accepted delta is in the 20–50% range or an absolute change of 5 mmol/l). Thus it is likely that in clinical practice, type 2 MI will be diagnosed with greater frequency and unstable angina will be less frequent.102
Myocardial injury is also likely to be diagnosed more frequently, with detection of more instances of non-ischaemic myocardial damage. In our opinion, it is foreseeable that myocardial injury may become a more common diagnosis than type 2 or type 1 MI in hospitalised patients.103 Patients with a non-coronary but otherwise cardiac cause of presentation to the emergency department (e.g. arrhythmia or myocarditis) may have higher hsTn levels than those with non-cardiac aetiologies, such as anaemia or hypoxia.104
HsTn might allow greater precision with risk stratification in certain clinical settings of type 2 MI, such as tachyarrhythmia. One study found that patients with tachyarrhythmia who had a positive hsTn (accounting for 47% of all tachyarrhythmia patients) had a significantly higher mortality than those with a negative hsTn, with rates similar to that of non-ST-elevation MI patients.80
Conclusion
Type 2 MI is common in hospital populations and accounts for at least 25% of all MI. Increased detection of both type 2 MI and myocardial injury with hsTn means that these entities will be frequently encountered by all clinicians. This is not surprising given the many clinical conditions that can disturb the supply–demand balance of oxygen to the myocardium and the many causes of myocardial injury. Patients with type 2 MI may be asymptomatic. This, combined with a lack of specific treatments for type 2 MI, can create some uncertainty for clinicians at the bedside. Most importantly, type 2 MI patients have a worse prognosis than type 1 MI patients.
Perhaps the most needed next step in the diagnosis of type 2 MI is a more specific clinical definition to streamline future investigation.25,27,33 In current clinical practice there are four main scenarios that are separated partly by their pathophysiology, but mainly by their investigative and management strategies: first, type 1 MI with an acute atherothrombotic event with plaque rupture and thrombus formation, for which PCI or thrombolytic therapy, dual antiplatelets, and anticoagulants are appropriate; second, type 2 MI with some degree of underlying CAD for which statin with or without aspirin may be appropriate, as well as β-blockers and ACEIs if the left ventricular ejection fraction is decreased, along with risk factor modification; third, type 2 MI due to supply–demand mismatch with minor coronary artery stenosis (i.e. MINOCA); and fourth, troponin elevation due to non-ischaemic mechanisms (e.g. myocardial injury).
Areas that need ongoing research so that they may be incorporated into a clinical universal definition are impact of hsTn assays; quantification of troponin levels and patterns; and specification of triggering factors for type 2 MI without being too exclusive (for example, the degree of anaemia, hypoxemia, hypertension, hypotension, tachypnoea, bradycardia etc.).32,33 Clearly this will be very challenging in the individual patient and confounded by multiple comorbidities. When there is a grey area, strategies to exclude plaque disruption and acute coronary thrombosis clinically with a certain degree of confidence would be very helpful. It must be remembered that all MIs involve thrombus in the coronary arteries post-mortem (Viramani, personal communication, 2021), and detection of plaque rupture is the critical feature.
Given the poor prognosis with type 2 MI, another main area of future investigation should be treatment and prevention. Tachycardia is a common final trigger in type 2 MI, and targeted heart rate reductions need to be evaluated, perhaps alongside such things as oxygen therapy and blood transfusions. The role of angiography and PCI needs to be further defined, and randomised trials are underway.101 The research effort afforded to type 1 MI over the last decade should be afforded to type 2 MI, with emphasis on medical therapies (such as those acting on ischaemic cellular signalling that have shown promise in non-human models) to achieve a reduction in morbidity and mortality. Long-term outcome reports of type 2 MI patients stratified by specific treatment strategy are needed. As well as treatment, the aim is to be able to identify patients at risk of type 2 MI and institute preventative strategies early.
In the current era of hsTn, further research and streamlined clinical guidelines are needed to provide greater awareness and confidence for clinicians in the prevention, diagnosis, risk stratification and management of type 2 MI, and with regard to the significant implications it has for future clinical outcomes.