










The discovery of proprotein convertase subtilisin/kexin type 9 (PCSK9) and its translation to an approved target of therapy in a span of a mere 12 years highlights how genetic insights can be leveraged into intelligent therapeutic innovation.1–3 The journey started in 2003, when Abifadel et al. discovered a cohort of French individuals with autosomal dominant hypercholesterolaemia, without mutations in the canonical familial hypercholesterolaemia genes (e.g. LDL receptor [LDLR] and apolipoprotein B100 [APOB]).4 It was later found that these individuals had a gain-of-function mutation in PCSK9, which is now understood as a key regulator in cholesterol homeostasis.4 As gain-of-function mutations in PCSK9 led to striking elevations in LDL cholesterol (LDL-C), it was conceivable that loss-of-function (LOF) mutations in PCSK9 would result in very low levels of LDL-C. Cohen and Hobbs investigated this hypothesis in the Dallas Heart Study by sequencing PCSK9 in those who had very low LDL-C, defined as <58 mg/dl.5 The investigators found that those with nonsense mutations (Y142X and C679X) leading to LOF of PCSK9 had an associated reduction of LDL-C by 40%.5 A similar study was performed in the ARIC cohort, where nonsense LOF mutations in PCSK9 (Y142X and C679X) were found in 2.6% of black participants, and sequence variations in PCSK9 (R46L) occurred in 3.2% of white participants, corresponding to LDL-C reductions of 28 and 15%, respectively.6 Additionally, these modest reductions in LDL-C translated to dramatic reductions in cardiovascular risk in black and white participants (HR 0.11; 95% CI [0.02–0.81] and HR 0.50; 95% CI [0.32–0.79]).6 Recently, protein-truncating variants in PCSK9 and APOB were assessed in 209,537 participants from five NHLBI cohorts and the UK Biobank.7 The prevalence of protein-truncating variants in either PCSK9 or APOB was 0.4%, and associated with a 45–49 mg/dl decrease in LDL-C, corresponding to a 49% lower risk in CHD.7 These results lend credence to the notion that cumulative lifelong exposure to LDL-C is a primary driver for atherosclerosis.6,8
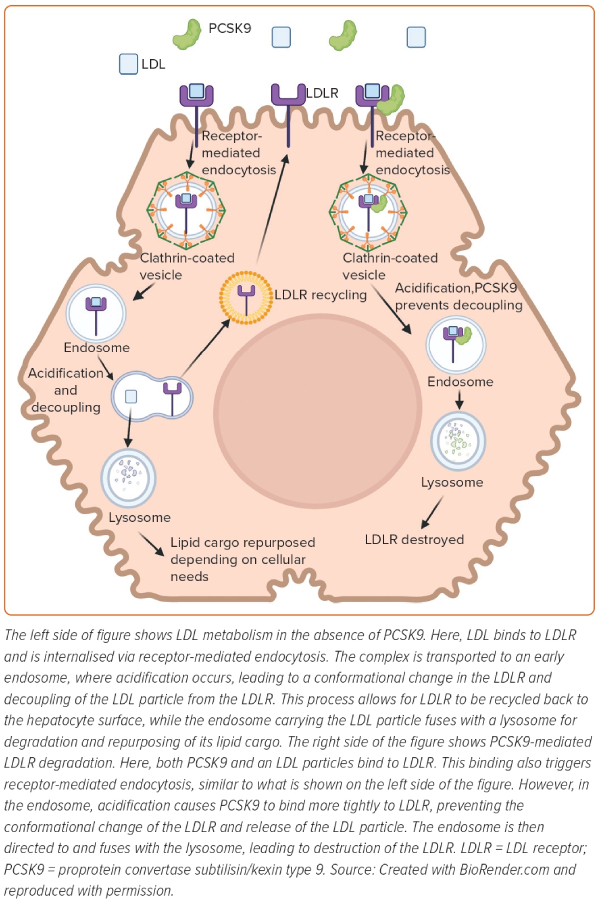
The majority of PCSK9 is secreted by the liver and impacts plasma LDL-C concentration by interfering with LDL receptor (LDLR) recycling. LDL binds to its receptor through the interaction of APOB on LDL with the LDLR on the hepatocyte surface (Figure 1).2 This binding leads to internalisation of LDL particles into the cell via a process known as receptor-mediated endocytosis.2 Once internalised, the LDL–LDLR complex is transported to an early endosome, which becomes increasingly acidic due to proton pumping by the voltage-dependent ATPase pump on the endosomal surface.2 The acidification of the late endosome leads to a conformation change in the receptor, which allows for the receptor to release the LDL particle.2 The endosome containing the LDL particle then fuses with the lysosome for degradation and repurposing of its lipid cargo, depending on cellular needs.2 In the meantime, the LDLR is recycled back to the surface of the cell to engage in further rounds of LDL clearance.2 The rapid recycling of LDLR during its 20-hour lifespan allows for clearance of approximately 100–150 LDL particles.2
As described below, PCSK9 interferes with the normal LDLR recycling loop and reduces the quantity of LDLRs at the hepatocyte surface. This action ultimately decreases the capacity of the hepatocytes to clear LDL from the circulation.5,9 In the endoplasmic reticulum, PCSK9 undergoes autocatalytic cleavage by means of its protease activity.5,9 After, PCSK9’s catalytic site is blocked by its cleaved prodomain, making it enzymatically inactive.10 PCSK9 is then secreted as a low-abundance plasma protein and non-enzymatically binds to LDLR at the epidermal growth factor precursor homology domain.11 With PCSK9 bound to LDLR, the process of receptor-mediated endocytosis continues in a normal fashion.12 However, once internalised, acidification in the endosome enhances the binding affinity of PCSK9 to LDLR.12 The strong binding of PCSK9 prevents the conformational change of LDLR, and thus, decoupling of LDL and LDLR does not occur.11,12 At this point, the endosome with its ternary complex (LDL–PCSK9–LDLR) fuses with the lysosome, and the LDLR is destroyed (Figure 1).10
Thus, gain-of-function mutations in PCSK9 reduce the density of LDLR on the cell surface and decrease the capacity to clear LDL from the plasma. Conversely, LOF mutations in PCSK9 lead to lower plasma LDL-C and profound cardiovascular protection.5 Additionally, those with homozygous LOF PCSK9 mutations have been shown to have lifelong severely low plasma LDL-C (<15 mg/dl), without health consequences.9 Although PCSK9 is expressed in several organs, including the liver, small intestine, brain and kidneys, the absence of circulating PCSK9 does not appear to impact cognition, physical development or reproductive capacity.9 In that regard, there has been intense interest in antagonising PCSK9 to increase LDLR recycling and enhance LDL particle clearance.13 The purpose of this review is to discuss current and emerging therapies targeted to PCSK9 to reduce LDL-C.
Fully Human Monoclonal Antibodies
Over the past four decades, there have been several advances in monoclonal antibody development.14 Initial monoclonal antibodies were fully mouse or chimeric, resulting in high immunogenicity, leading to limited efficacy and safety concerns.14 However, with advances in recombinant DNA technology, fully human monoclonal antibodies have been developed that do not elicit human antimouse antibodies; and thus, are not rapidly cleared, resulting in greater efficacy and fewer hypersensitivity reactions.14 Monoclonal antibodies are advantageous in that they bypass liver and kidney metabolism, minimising drug interactions, and are specific in binding to targets of interest via the antigen binding site, rendering these targets ineffective and minimising off-site effects.14 Thus, with advances in monoclonal technology, there was an enticing rationale to use fully human monoclonal antibodies to inhibit PCSK9.15
Initial Phase I and II trials of fully human monoclonal antibodies targeting PCSK9 were studied as monotherapy, in combination with other lipid-lowering therapies, and in various populations, including healthy volunteers and patients with hypercholesterolaemia.15 Overall, these studies revealed drastic LDL-C reductions of approximately 50–60% compared with placebo with favourable side-effect profiles, aside from mild injection site reactions.15 Given these positive findings, evolocumab and alirocumab, both fully human monoclonal antibodies that target extracellular PCSK9, were studied in two major Phase III cardiovascular outcome trials: FOURIER and ODYSSEY OUTCOMES.16,17
In FOURIER, 27,564 participants with a history of MI, stroke or symptomatic peripheral arterial disease on maximally tolerated statin therapy and LDL-C ≥70 mg/dl were randomised to evolocumab versus placebo.16 There was a statistically significant reduction in cardiovascular events in those receiving evolocumab compared with placebo, 9.8 versus 11.3%, respectively, over a median follow-up of 2.2 years.16
Long-term safety and efficacy of evolocumab was recently demonstrated in FOURIER-OLE, which included 6,635 participants from the main FOURIER trial.18 Individuals who received evolocumab sustained 15% lower risk of major adverse cardiovascular events compared with those who received placebo, over an average follow-up of 7.1 years (HR 0.85; 95% CI [0.75–0.96]).18 Additionally, long-term safety data from FOURIER-OLE were comparable between the evolocumab and placebo groups, aside from slightly higher injection site reactions in the evolocumab group.18 Importantly, FOURIER-OLE included participants with the longest exposure to PCSK9 inhibitor thus far.18 The findings demonstrate continued safety and tolerability of longer-term dramatic LDL-C reduction with evolocumab.18 This is significant, as there has been concern regarding possible harmful effects from very low LDL-C, such as diabetes, stroke, cataracts and neurocognitive impairment.19 However, majority of studies argue against these harmful complications from very low LDL-C values.19 In fact, in a subgroup analysis from FOURIER, cognitive testing was performed and there was no difference in cognitive function in those who received evolocumab compared with placebo.20 Moreover, participants who received evolocumab both in the main FOURIER trial and in FOURIER-OLE demonstrated better atherosclerotic cardiovascular disease (ASCVD) outcomes.18 The findings are important because they add to the mounting evidence that indicates that the benefits of LDL-C lowering accumulate with time and that starting LDL-lowering earlier is more effective at preventing events.21
In ODYSSEY OUTCOMES, 18,924 individuals who had a recent acute coronary syndrome within the past 1–12 months were randomised to treatment with alirocumab or placebo every 2 weeks.17 All participants were on maximally tolerated statin therapy for at least 2 weeks prior to randomisation and had LDL-C ≥70 mg/dl, non-HDL-C ≥100 mg/dl or APOB ≥80 mg/dl.17 There was a statistically significant reduction in recurrent major adverse cardiovascular events in those receiving alirocumab compared with placebo, 9.5 and 11.1%, respectively, over a median follow-up of 2.8 years.17 When the population was stratified by LDL-C values (<80 mg/dl, 80–<100 mg/dl and ≥100 mg/dl), individuals with LDL-C ≥100 mg/dl garnered the greatest benefit from alirocumab compared with placebo for any cardiovascular event (HR 0.78; 95% CI [0.69–0.89]).17
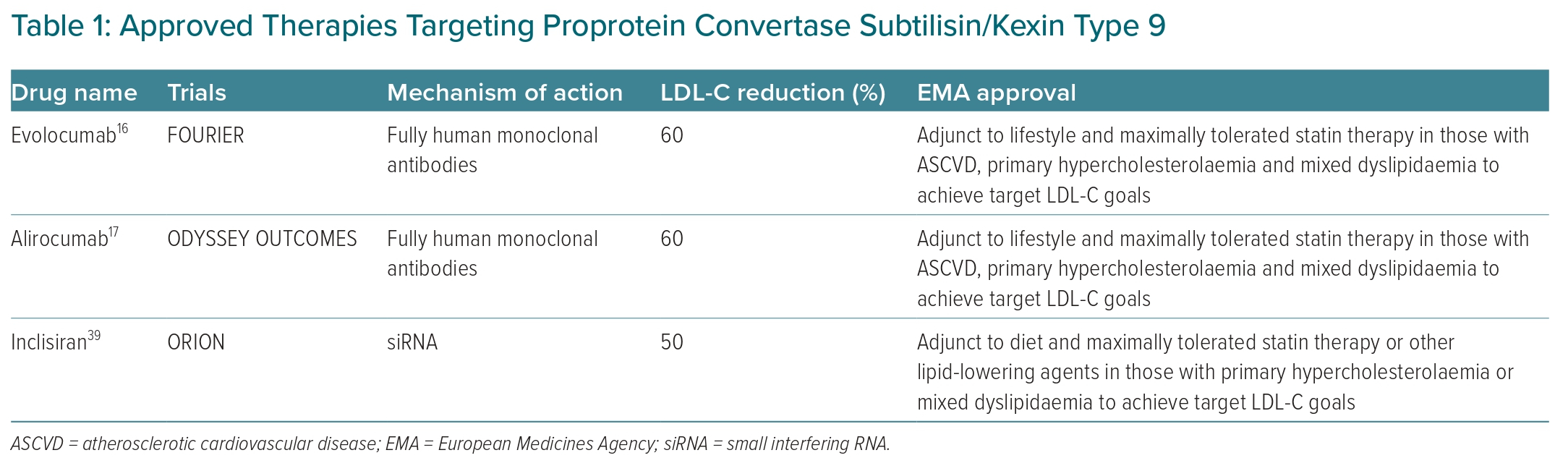
The above trials illustrate a clear cardiovascular benefit from monoclonal antibodies targeting PCSK9.16–18 It is important to note that these trials were in individuals with existing cardiovascular disease and on maximally tolerated statin therapy at baseline.16,17 Based on the results of the PCSK9 inhibitor cardiovascular outcomes trials, the European Medicines Agency approved alirocumab and evolocumab as an adjunct to lifestyle and maximally tolerated statin therapy for those with ASCVD, primary hypercholesterolaemia or mixed dyslipidaemia to achieve target LDL-C goals (Table 1).22 Additionally, VESALIUS-CV (NCT03872401), an ongoing phase III randomised controlled trial of >12,000 participants, is investigating the impact of evolocumab on incident major cardiovascular events in those who are at high cardiovascular risk, but without a history of MI or stroke, over a follow-up period of at least 4.5 years. If the results of VESALIUS-CV are positive, it has the potential to expand the indication for monoclonal antibodies inhibiting PCSK9.
In addition to evaluating the role of PCSK9 inhibition on LDL-C reduction and ASCVD outcomes, the aforementioned trials provide insight into the relationship between lipoprotein(a) (Lp[a]) and PCSK9. Lp(a) is an atherogenic, antifibrinolytic, pro-inflammatory LDL-like particle with an APOB and apolipoprotein(a) component.23 Lp(a) levels are largely genetically determined and independently associated with ASCVD.23 Interestingly, monoclonal antibodies targeting PCSK9 do not just lower LDL-C by 60%, but also lower Lp(a) by approximately 25%.17,24
In an analysis from FOURIER, evolocumab reduced Lp(a) by 26.9%, and the study found that those with elevated Lp(a) at baseline garnered a greater cardiovascular benefit with evolocumab versus placebo compared with those without elevated Lp(a) (HR 0.77; 95% CI [0.67–0.88] and HR 0.93; 95% CI [0.80–1.08]), respectively.25
In a secondary analysis of ODYSSEY OUTCOMES, it was shown that the benefit of alirocumab in those with LDL-C near 70 mg/dl was only present when Lp(a) was elevated, suggesting that the cardiovascular benefit associated with alirocumab may partly be due to concomitant Lp(a) reduction, not simply LDL-C lowering.24 Although these findings may suggest that the impact of PCSK9 inhibitors on Lp(a) may be LDLR mediated, other LDLR-dependent lipid-lowering drugs, such as statins and ezetimibe, do not impact Lp(a).26,27 Moreover, Shapiro et al. demonstrated a high proportion of discordant LDL-C/Lp(a) responses to PCSK9 inhibition.28 This consistent finding indicates that Lp(a) may not always or exclusively use LDLR as a clearance receptor, and thus, there likely are other mechanisms by which PCSK9 impacts Lp(a) clearance.27,28
Small Interfering RNA
Inclisiran is a small interfering RNA (siRNA) therapy consisting of a synthetic nucleotide sense strand, as well as an antisense strand, which are conjugated to the N-acetylgalactosamine (GalNAc) ligand.29–32 GalNAc binds to the asialoglycoprotein receptor, exclusively expressed on the sinusoidal surface of the liver.29,30,32 Thus, conjugation of the nucleotide strands to GalNAc effectively targets inclisiran to the liver, minimising off-target effects and lowering the dose that needs to be administered.32,33 Once inside the cell cytoplasm, the antisense strand is loaded into an RNA-induced silencing complex.31,32 The loaded RNA-induced silencing complex then binds to PCSK9 messenger RNA (mRNA), leading to its degradation and preventing protein translation.29,30,34
The clinical development of inclisiran has proceeded through the ORION program. These trials have evaluated the safety and efficacy of inclisiran. In the Phase II multiple ascending dose trial, ORION-1, participants at high risk for ASCVD with elevated LDL-C were randomly assigned to receive one dose of placebo or 200/300/500 mg of inclisiran or two doses (at days 1 and 90) of placebo or 100/200/300 mg of inclisiran.35 The primary endpoint was LDL-C reduction at 180 days. The investigators found that the optimal dose for LDL-C lowering with inclisiran was 300 mg given twice as starting regimen. All patients responded with significant LDL-C lowering. At 6 months, the mean LDL-C and PCSK9 reduction in those who received two doses of 300 mg inclisiran compared with placebo was 54.4 and 67.9%, respectively. There were no safety concerns. These changes were largely sustained over a longer study duration in the Phase II open-label non-randomised extension, ORION-3, where mean LDL-C reduction over 4 years in those treated with inclisiran was 44.2% (95% CI [41.4–47.1]).36
Three Phase III randomised, placebo-controlled LDL-C-lowering efficacy trials were performed and designed with similar methodology with a prespecified intention to pool the data at the completion of the studies, ORION-9, 10 and 11.37,38 Patient populations were different in the three trials: ORION-9 included individuals with heterozygous familial hypercholesterolaemia and LDL-C ≥100 mg/dl; ORION-10 included individuals from the US with a history of ASCVD and LDL-C ≥70 mg/dl; and ORION-11 included participants from South Africa and Europe with a history of ASCVD and LDL-C ≥70 mg/dl or an ASCVD risk equivalent and LDL-C≥100 mg/dl.37,38 ASCVD risk equivalents in ORION-11 were defined as those with type 2 diabetes, familial hypercholesterolaemia or a 10-year cardiovascular disease risk ≥20%.38 In all of these Phase III trials, participants were required to be on maximally tolerated lipid-lowering therapy for at least 30 days prior to randomisation to placebo or inclisiran.9 Dosing was done at day 1, 90 and 6 months thereafter using 300 mg of inclisiran sodium or placebo. All trials had an end time of 540 days, which was 90 days after the fourth and final inclisiran dose. In the pooled patient-level analysis of these trials, including 3,660 individuals, the placebo-corrected LDL-C reduction was 50.7% (95% CI [48.4–52.9]).39 Safety events were similar in the pooled analysis, aside from higher injection site reactions in the inclisiran group. The vast majority of the injection site reactions were mild.
A recent analysis of the potential cardiovascular benefit from inclisiran was completed by Ray et al., using non-adjudicated prespecified and non-prespecified cardiovascular outcome exploratory endpoints from pooled patient-level data of the three Phase III inclisiran trials.40 The prespecified exploratory endpoint was a composite of cardiac death, cardiac arrest, non-fatal MI, and fatal and non-fatal stroke. Non-prespecified endpoints included fatal and non-fatal MI, and fatal and non-fatal stroke. Using this prespecified exploratory endpoint, individuals in the inclisiran group showed cardiovascular benefit relative to participants allocated to placebo (OR 0.74; CI 95% [0.58–0.94]). The point estimates of the non-prespecified exploratory endpoints favoured a cardiovascular benefit from inclisiran compared with placebo, but the confidence intervals crossed one (OR 0.8; CI 95% [0.50–1.27]) and (OR 0.86; CI 95% [0.41–1.81]) for fatal and non-fatal MI, and fatal and non-fatal stroke, respectively. It is important to recognise that this analysis was exploratory, and none of the pooled Phase III LDL-C-lowering efficacy trials were powered to assess cardiovascular outcomes.
More importantly, there are two large randomised controlled cardiovascular outcome trials under way testing the cardiovascular efficacy of inclisiran, ORION-4 and VICTORION-2 PREVENT. Both studies will assess the impact of inclisiran on major adverse cardiovascular events in a combined total of approximately 30,000 participants with a history of ASCVD. ORION-4 (NCT03705234) and VICTORION-2 PREVENT (NCT05030428) have planned completion dates of 2026 and 2027, respectively. On 9 December 2020, the European Medicines Agency approved inclisiran for use in individuals with primary hypercholesterolaemia or mixed dyslipidaemia as an adjunct treatment when LDL-C goals are not met through diet, maximally tolerated statin therapy or other lipid-lowering agents (Table 1).41
Small Oral Molecules
An attractive strategy to inhibit PCSK9 is with oral delivery, reducing costs and the need for subcutaneous injections.13 Development of oral PCSK9 inhibitors poses several technical challenges, given the planar structure of the catalytic domain of PCSK9.13 However, with advances in medicinal chemistry, several oral PCSK9 inhibitors are in clinical development. One such oral molecule is MK-0616, a macrocyclic peptide that was found to lower PCSK9 by >90% compared with baseline in a Phase I cross-over design trial of 60 healthy men.42 In another Phase I trial of 40 men and women, MK-0616 was shown to reduce LDL-C by 65% compared with baseline after 14 days of therapy.42 A Phase IIb, randomised, double-blind, placebo-controlled trial studying the efficacy of MK-0616 recently completed enrolment (NCT05261126; Table 2).
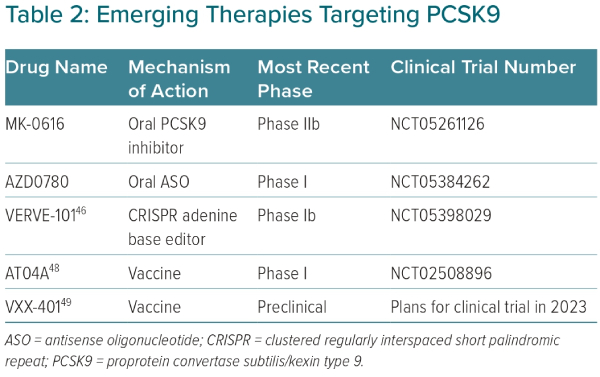
Another emerging strategy to inhibit PCSK9 involves an oral antisense oligonucleotide (ASO).34,43 Similar to siRNA therapies, ASOs silence gene expression, preventing protein translation.34 However, unlike siRNA therapies, ASOs are single-stranded nucleotides that enter the cell’s nucleus to target complementary mRNA. Once the ASO binds to the complementary mRNA, ribonuclease H becomes activated, leading to destruction of the target mRNA. Until now, ASO therapies have used injectable deliveries. Oral ASO delivery poses several challenges, as these compounds are hydrophilic, must pass through the acidic gastric environment and are not readily absorbed through the intestinal system.43 Thus, several chemical modifications are needed to increase compound stability, intestinal permeability and potency while minimising off-target effects. In a preclinical study with dogs, an oral enteric-coated ASO, modified with sodium caprate to increase intestinal permeability, was shown to result in clinically meaningful liver bioavailability.43 Given these promising preclinical findings, an actively recruiting Phase I trial of an oral ASO, AZD0780, is under way to assess safety and tolerability in humans (NCT05384262; Table 2). Several other oral methods to target PCSK9 are currently in development.13
Clustered Regularly Interspaced Short Palindromic Repeat
A novel method of permanently silencing hepatic PCSK9 with a single infusion involves in vivo gene editing with clustered regularly interspaced short palindromic repeat (CRISPR) technology.44 Compared with standard CRISPR gene editing nucleases, CRISPR DNA base editing does not require double strand breaks, but rather, swaps a single nucleotide, allowing for safer and more precise gene silencing.44,45 Treatment with CRISPR DNA base editing has been shown to be effective in silencing PCSK9, with VERVE-101 furthest in development.46 VERVE-101 is a CRISPR adenine base editor composed of an mRNA and a guide RNA strand contained in a GalNAc-lipid nanoparticle.45 The GalNAc-lipid nanoparticle enhances hepatic delivery, and once inside the hepatocyte, the mRNA and guide RNA strands are released in the cytosol.45,46 The mRNA strand is translated to an adenine base editor, which subsequently binds to the guide RNA strand.45,46 The complex then localises to the nucleus, where the guide RNA strand identifies PCSK9, allowing for the adenine base editor to make a single adenine-to-guanine nucleotide exchange, essentially causing a LOF mutation and permanently silencing PCSK9.45
In a study of cynomolgus monkeys, a single dose of VERVE-101 was able to reduce PCSK9 levels by 90% and LDL-C by 60%, with stable values over the next 8 months.44 Recently, the long-term effects of VERVE-101 were studied in 36 non-human primates at placebo, low dose (0.75 mg/kg) or high dose (1.5 mg/kg).45 After 476 days of follow-up, the highest dose of VERVE-101 reduced PCSK9 by 83% and LDL-C by 69%. Most importantly, there were no concerning long-term safety effects of VERVE-101. There were slight increases in liver transaminases, but these elevations resolved after 14 days from time of initial infusion. Additionally, there were no pathological macroscopic or histological changes due to VERVE-101 in biopsied organs. Finally, in a study to assess germline editing, VERVE-101 demonstrated no editing of PCSK9 in sperm samples obtained 90 days after dosing.45 The promising safety and efficacy of VERVE-101 in non-human primates has given rise to an open-label, single ascending Phase Ib clinical trial, with the first human dose given on 12 July 2022 (NCT05398029; Table 2).45,46
Anti-PCSK9 Vaccine
While passive vaccination occurs with anti-PCSK9 antibodies (alirocumab, evolocumab), treatment with an active vaccination may offer advantages in terms of dosing, adherence and cost.47 The development of anti-PCSK9 peptide vaccines is an arduous task, where a balance is needed between eliciting a strong antibody response against PCSK9, while also limiting off-target effects.48 AT04A is an anti-PCSK9 peptide-based vaccine that recently completed a Phase I trial and was found to be safe with statistically significant LDL-C lowering of 7.2% compared with placebo.48 Although overall a modest LDL-C reduction, those with the highest PCSK9-specific antibody titers had approximately 40% LDL-C-lowering compared with placebo. Thus, AT04A is undergoing further development to enhance immunogenicity and potentially further reduce LDL-C (Table 2). Another peptide-based vaccine in development, VXX-401, recently completed two preclinical studies in non-human primates and was found to be safe with sustained LDL-C reductions compared with placebo by approximately 50%.49 VXX-401 will progress to clinical trials later in 2023 (Table 2).
Clinical Use
Current approved therapies targeting PCSK9 include evolocumab, alirocumab and inclisiran.50,51 The joint 2019 European Society of Cardiology and European Atherosclerosis Society guidelines recommend the use of evolocumab and alirocumab in those with ASCVD or high-risk primary prevention patients who are unable to achieve an LDL-C reduction of at least 50% and an LDL-C value of <55 mg/dl on maximally tolerated statin therapy in combination with ezetimibe.50 High-risk primary prevention includes those with type 2 diabetes and target organ damage, long-term type 1 diabetes, severe chronic kidney disease, familial hypercholesterolaemia and another cardiovascular risk factor, or a calculated systematic coronary risk estimation score of at least 10%. Additionally, inclisiran has been approved by the European Medicines Agency and can be used in the background of maximally tolerated lipid-lowering therapy to attain LDL-C goals.51
One of the greatest barriers to prescribing evolocumab and alirocumab is cost. Initially, these therapies were greater than $14,000 a year, but have decreased over time to approximately $6,000 a year.52 Multiple cost analyses have shown that at this current price, evolocumab and alirocumab are high value for those at greatest ASCVD risk.52 However, as prices continue to decline, these therapies will likely be cost-effective for lower-risk populations who are unable to meet LDL-C goals on maximally tolerated statin and ezetimibe therapy. While just recently brought to the market, inclisiran is approximately $6,500 a year, and its cost-effectiveness is yet to be determined.53
Conclusion
The landscape of cholesterol management has forever been revolutionised by the discovery of PCSK9. From genetic studies to randomised controlled trials, PCSK9 is now understood to be a major player in cholesterol homeostasis, specifically relating to its impact on LDLR recycling.2 Clinical trials testing fully human monoclonal antibodies demonstrated that blocking extracellular PCSK9 leads to significant reductions in both plasma LDL-C and cardiovascular risk, which remains stable over long-term follow-up.16–18 In addition, siRNA therapy with inclisiran has shown excellent LDL-C-lowering efficacy, with cardiovascular outcome trials under way.31 Other mechanisms of targeting PCSK9, including small oral molecules, CRISPR and vaccines, are in development.13 These advances have the potential to expand access to therapies targeting PCSK9 by reducing cost, limiting need for subcutaneous injections and/or limiting the quantity of subcutaneous injections. Future research will continue to evaluate the safety and efficacy of novel PCSK9 therapies, and define which population reaps the greatest cardiovascular benefit from treatment. Without a doubt, the PCSK9 saga continues with realistic hopes of further reducing the burden of cardiovascular disease. 