







Acute coronary syndromes (ACS) comprise a major cause of morbidity and mortality. The almost-exclusive cause of ACS, including unstable angina, acute myocardial infarction (AMI) with or without ST-segment elevation, and sudden cardiac death, is atherothrombosis. Pilot pathological studies have identified the pathophysiological processes implicated in the destabilisation and thrombosis of atheromatous plaques.1–3 According to the current understanding of ACS, plaques with specific morphological characteristics are more prone to rupture and development of thrombosis, which subsequently leads to the clinical manifestation of ACS. However, pathological studies are limited by their cross-sectional and teleological nature, thus imaging tools have been developed that can assess such morphological characteristics in vivo. Intracoronary imaging techniques can assess coronary plaques beyond luminal assessment (i.e. coronary angiography) and can visualise atheromatous plaques in vivo.4 Among these techniques, due to its high resolution (~15 μm) optical coherence tomography (OCT) can identify the majority of the morphological characteristics of stable and complicated atheromatous plaques, including precise measurement of the thickness of the overlying fibrous plaque and a high sensitivity for thrombus detection.5 Thus, OCT offers a valuable tool for obtaining in vivo insights into the pathomechanisms of ACS.6
Pathological Paradigm of the Vulnerable Plaque
Post-mortem studies of sudden cardiac death victims have shown that the major pathological substrates of coronary thrombosis are plaque rupture in 60–70 % of cases, plaque erosion in 20–30 % and thrombosis triggered by calcified nodules in 2–5 %.1–3 Plaque rupture, the most frequent mechanism of coronary thrombosis, involves disruption of a thin fibrous cap that overlies a large necrotic core, causing the thrombogenic contents of the necrotic core to come into contact with the bloodstream and trigger thrombus formation. Plaque erosion is defined as the presence of thrombosis in the absence of plaque thrombosis, often with absence of an endothelial layer at the thrombosed sites.2 Calcified nodules is a less common cause of coronary thrombosis, characterised by thrombus formation over nodular calcification protruding into the lumen through a disrupted thin fibrous cap.
Plaques that are at high risk of undergoing acute thrombosis are called vulnerable plaques and bear morphological resemblance to the aforementioned plaques.7 Nevertheless, eroded plaques do not have specific distinct morphological characteristics; therefore, the identification of erosion-prone plaques is challenging. In contrast to eroded plaques, plaques undergoing rupture have distinct morphological characteristics, including a large necrotic core, a thin (<65 μm in pathology series) and inflamed fibrous cap, high plaque burden, intimal vascularisation, inflammatory cell infiltration and spotty calcification.8 These characteristics are also observed in another type of plaque, called thin cap fibroatheroma (TCFA), which is considered the precursor of the ruptured plaque, and thus the main morphological subtype of vulnerable plaques.8
Optical Coherence Tomography Features of Stable Coronary Plaques
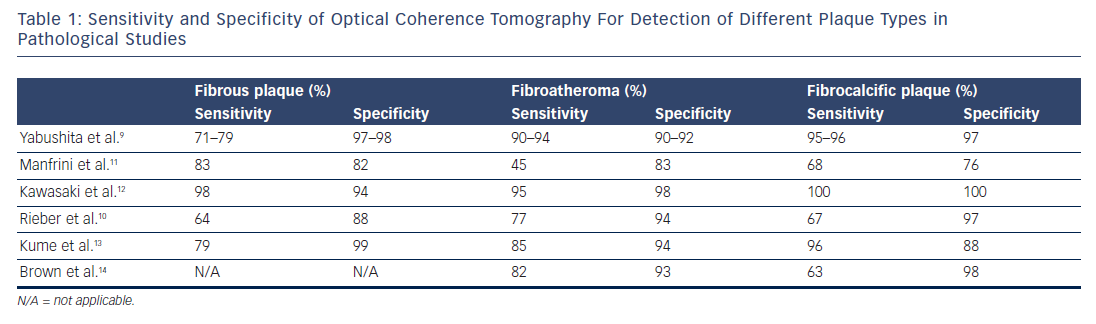
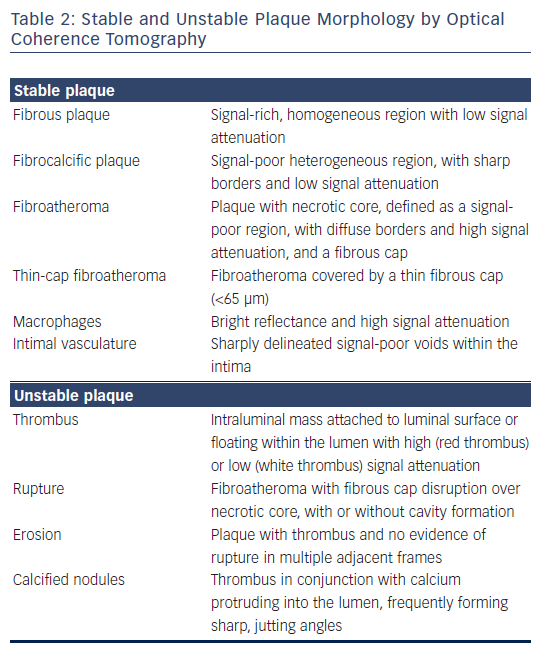
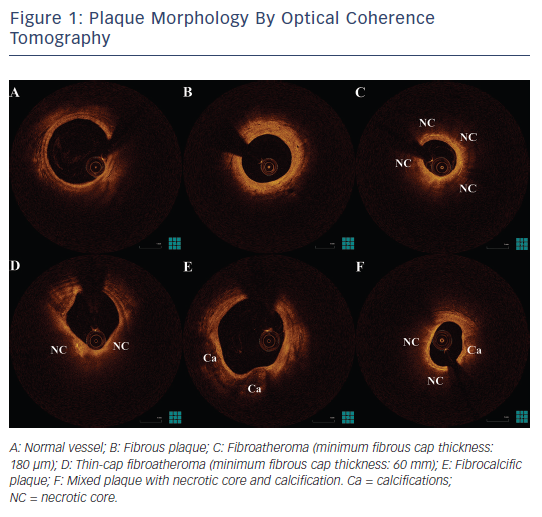
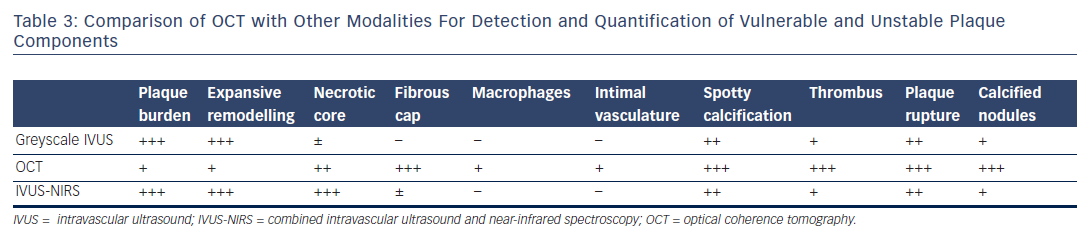
Post-mortem OCT studies have defined the optical appearance and morphological characterisation of the plaque,9–14 showing an overall good agreement with histology and high reproducibility (see Table 1). OCT can identify normal wall segments and early atherosclerotic lesions,15,16 and also perform tissue characterisation in advanced atherosclerotic lesions (see Table 2).5 The major plaque types identified by OCT are fibrous plaques, appearing as a signal-rich, homogeneous region with low signal attenuation; fibrocalcific plaques, appearing as a signal-poor heterogeneous region, with sharp borders and low signal attenuation; and necrotic core, which appears as a signal-poor region, with diffuse borders and high signal attenuation within a lesion that is covered by a fibrous cap.5,9 Fibroatheroma has been defined as a plaque with a necrotic core and a fibrous cap. Importantly, the thickness of the fibrous cap can be precisely measured by OCT.17,18 A thin-cap fibroatheroma identified by OCT is defined as a fibroatheroma covered by a thin fibrous cap; the current threshold of thin cap is the extrapolated by histology value of 65 μm, although the in vivo optimal threshold is controversial. Mixed plaques identified by OCT are those with more than one plaque morphology within a single cross-sectional frame. Additional tissue components that can be identified by OCT include macrophages that are characterised by bright reflectance and high signal attenuation, and intimal vasculature that appears as sharply delineated signal-poor voids within the intima.5,19Figure 1 demonstrates the main plaque types by OCT and a comparison of OCT with other invasive imaging modalities is presented in Table 3.
Optical Coherence Tomography Features of Unstable Coronary Plaques
Importantly, OCT can also be used for tissue characterisation in thrombosed plaques by imaging thrombus as an intraluminal mass attached to the luminal surface or floating within the lumen.20 Plaque rupture can be identified by OCT as a fibroatheroma with fibrous cap disruption over necrotic core, with or without cavity formation.5 While plaque erosion is defined as a plaque with thrombus and no evidence of rupture in multiple adjacent frames, although due to the presence of red thrombus, plaque rupture can be obscured, which can lead to an underestimation of its incidence in lesions with red thrombus. Thrombosis of calcified nodules can be identified as evidence of thrombus in conjunction with calcium protruding into the lumen, frequently forming sharp, jutting angles.21
Plaque Morphology and Clinical Presentation
As OCT can accurately image thrombosed plaques, a number of OCT studies have focused on examining pathophysiological aspects of vulnerable plaques, including mechanisms of plaque formation, progression and destabilisation. It has been shown by OCT that vulnerable plaque morphology with thin fibrous cap and OCT-detected TCFA is more frequent in unstable angina, non-ST-segment elevation myocardial infarction (NSTEMI), or ST-segment elevation myocardial infarction (STEMI) compared with stable angina.22,23 Moreover, OCT can more accurately assess complicated plaque morphology and thrombus in ACS compared with intravascular ultrasound (IVUS) or coronary angioscopy,24 enabling the evaluation of the incidence of pathomechanisms of ACS in an in vivo setting.25 In an in vivo OCT study of ACS, plaque rupture was identified in the culprit lesion of 44 % of the patients, erosion in 31 %, thrombosis of calcified nodules in 8 %, and other causes (including subocclusive stenosis, dissection and coronary spasm) in 17 %,26 showing a discordance with post-mortem studies. Of note, plaque rupture was more common in STEMI, in contrast to erosion, which was more common in patients with non-ST elevation ACS and calcified nodules that were exclusively observed in non-ST elevation ACS.
Differences in morphological characteristics of the ruptured plaque also translate into differences in clinical presentation. Ruptured plaques of patients with STEMI are associated with a greater extent of cap disruption and a smaller minimal lumen area compared with ruptured plaques of patients with NSTEMI.27,28 Similarly, ruptured culprit plaques of ACS had a lower lumen area in comparison with non-culprit plaques in the same patients who had undergone silent rupture.29 Moreover, asymptomatic patients with silent plaque rupture on OCT examination had a higher minimal lumen area than patients with non-ST-segment elevation ACS with culprit lesion plaque rupture.30 Collectively, these studies provide indirect evidence that the pre-existing lumen narrowing might also contribute to the clinical manifestation following plaque rupture. Morphological differences in ruptured plaques have also been found between ruptured culprit plaques of patients with exercise-triggered ACS and ruptured culprit plaques of patients with rest-onset ACS, where patients with exercisetriggered ACS had a higher cap thickness and higher incidence of rupture at the shoulder of the plaque.31 Finally, the presence of culprit lesion rupture in patients with ACS has been associated with lower incidence of prodromal symptoms compared with patients without evidence of culprit plaque rupture.32
Morphology of the Vulnerable Plaque: In Vivo Measurement of Fibrous Cap
OCT has shown an association of positive remodelling of the culprit lesion with TCFA morphology and inflammation,33,34 both being important features of the vulnerable plaque as shown in pathological and in vivo IVUS studies.8,35 Furthermore, OCT has been used for identifying a critical value of cap thickness that characterises ruptureprone plaques in vivo. Although autopsy series have shown that ruptured caps have a mean thickness of 23 ± 19 μm, with 95 % being thinner than 64 μm,1 the overestimation of cap thickness by OCT in comparison with histology17 and the effect of anisotropic tissue shrinkage in histology18,36 indicate that these thresholds might not be applicable in an in vivo setting. OCT studies of ruptured plaques have shown that cap thickness in ruptured plaques can be as high as 160 μm,27,31 and it is consistently thinner in the rupture site compared with the minimal lumen area site.27 The current consensus for defining thin cap by OCT is using the histology-derived threshold of 65 μm. However, it has been shown that a fibrous cap thickness of 67 μm has only 83 % sensitivity and 77 % specificity for discriminating between ruptured and non-ruptured plaques in vivo.37 Therefore, not only fibroatheromas with cap thickness <65 μm, but also those with cap thickness ≤100 μm, could be prone to rupture.
Localisation of Vulnerable Plaques
Knowledge of spatial distribution of vulnerable plaques may lead to more effective screening of these plaques. Angiographic studies have shown that most STEMI events tend to cluster within the proximal portion of coronary artery,38 matching the distribution of coronary atherosclerosis.39 Similarly, thin-cap fibroatheromas and ruptured plaques are localised in focal spots within the proximal third of coronary art, as shown in pathological studies.40,41 OCT studies have also tried to assess plaque morphology in relation to plaque localisation. OCT can be used for assessment of plaque morphology in all coronary vessels including left main lesions,42 although imaging of true ostial lesions (i.e. ostial left main and ostial right coronary artery) might be more challenging and require more imaging attempts also with withdrawal of the guiding catheter to the aorta.43–46 Preliminary OCT studies have also demonstrated a localisation of TCFAs at the proximal segment of coronary arteries,31,47 while our group has further demonstrated that culprit lesions of patients with ACS located in the proximal 30 mm of coronary arteries were more often associated with rupture and TCFA morphology compared with the more distal lesions, indicating that plaque rupture is the main pathomechanism for ACS in these proximal lesions.48 This knowledge of the spatial distribution of the high-risk plaques can be potentially useful in the future for the development of preventive treatment strategies.49–51
Multiple Coronary Lesion Instability in Acute Coronary Syndromes
Although ACS are in principle focal manifestations arising from a single lesion, multiple lesion instability is commonly observed in atherothrombosis.52–54 Thus, non-culprit lesions of patients with MI have a more complex angiographic morphology and are associated with rapid lesion progression and increased event rates at follow-up.55,56 In addition, patients with STEMI undergoing primary percutaneous coronary intervention of the culprit lesion with additional prophylactic revascularisation of non-culprit lesion with >50 % stenosis had fewer events than patients with culprit-only treatment.57 Based on these observations, it has been hypothesised that non-culprit lesions of patients with ACS have more unstable plaque morphology than lesions of stable patients. OCT studies have further shown increased incidence of non-culprit lesion TCFA and thrombus, and a higher incidence of multiple TCFA lesions in non-culprit lesions of patients with AMI compared with non-culprit lesions of patients with stable angina.23,58 Furthermore, non-culprit lesions of patients with AMI have a higher lipid content, a thinner fibrous cap and a higher incidence of macrophage infiltration and thrombus, in conjunction with higher incidence of TCFA.59 This is mainly observed in patients with rupture of the culprit plaque as pathomechanism for the ACS,60 and is more pronounced in patients with diabetes, in whom the incidence of non-culprit lesion TCFA and rupture is higher than in those without diabetes.61
Vulnerable Plaques and Thrombolytic Therapy
In OCT studies, vulnerable plaque morphology has also been associated with impaired flow after thrombolytic therapy administration, where increased lipid content, thinner fibrous cap and plaque rupture were more common among patients without complete flow restoration compared with those with complete flow restoration. Although the reason behind this observation is not entirely clear, increased thrombogenicity of vulnerable plaques due to an exaggerated inflammatory reaction has been speculated to play an important role.62 Furthermore, in patients with STEMI with a ruptured culprit lesion, the residual thrombus burden by OCT 1 day after fibrinolysis was higher than in patients with erosion of the culprit lesion, with the presence of white thrombus at the site of the rupture,63 thus underscoring the importance of the underlying plaque morphology in STEMI as a major determinant of the response to thrombolytic therapy.
Neoatherosclerosis and the Vulnerable Plaque
The long-term follow-up of both bare metal and drug-eluting stents has demonstrated that these devices are subject to failure, and have long intervals after implantation (up to 15–20 years).64–66 Pathological studies together with in vivo OCT observations have demonstrated a variety in the neointimal patterns observed at follow-up after stent implantation, which resemble components of native atherosclerosis, and imply the atherosclerotic change of the neointimal tissue.67–71 Neoatherosclerosis has been defined by pathological studies as the presence of clusters of lipid-laden foamy macrophages with or without necrotic core formation, and/or calcification within the neointimal tissue of stented segments.68,72 Several OCT studies have demonstrated the association of neoatherosclerosis development with late stent failure.72 The role of neointimal disruption, analogue to plaque rupture in native atherosclerosis, to very late stent thrombosis is under investigation.64 Neointimal rupture has been shown to be a very prevalent mechanism in very late bare metal stent thrombosis, especially a long time after implantation,73 and is one of the most common causes of late and very late drug-eluting stent thrombosis.66,74 Neointimal rupture is a mechanism occurring mainly at longer intervals following stent implantation,75 in regions with high necrotic core content and thin fibrous cap76 (similarly to what is observed with vulnerable plaques in native atherosclerosis) and a high incidence of macrophage infiltration.76 These features are being identified mainly by OCT.72
Shortcomings of Current Vulnerable Plaque Detection Strategies: New Targets For Imaging
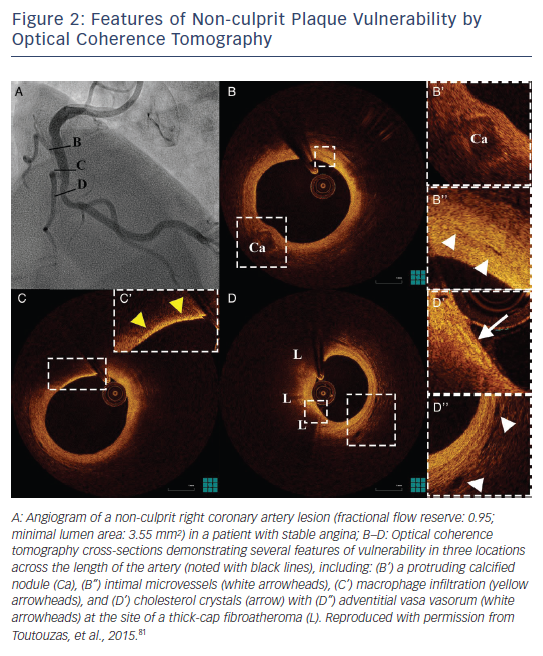
Although vulnerable plaque morphology has been prospectively associated with adverse outcomes in studies of IVUS with virtual histology and near-infrared spectroscopy, the predictive value of plaque morphology is poor, due to a high prevalence of these findings and a relatively low event rate.77–80 Consequently, despite the prognostic value of TCFA, the need to identify patients who are at high risk of cardiovascular events remains unmet.81 Therefore, new imaging targets that can enhance the prognostic potential of invasive imaging for lesion-level future events need to be identified. The identification by OCT of new imaging targets such as calcified nodules, intimal vasculature, macrophages and cholesterol crystals can offer additional information over the vulnerability of atheromatous plaques (see Figure 2). Moreover, the combination of anatomical assessment by OCT and biomechanical assessment of endothelial shear stress could give further insight into high-risk plaques.82–84
Future Developments
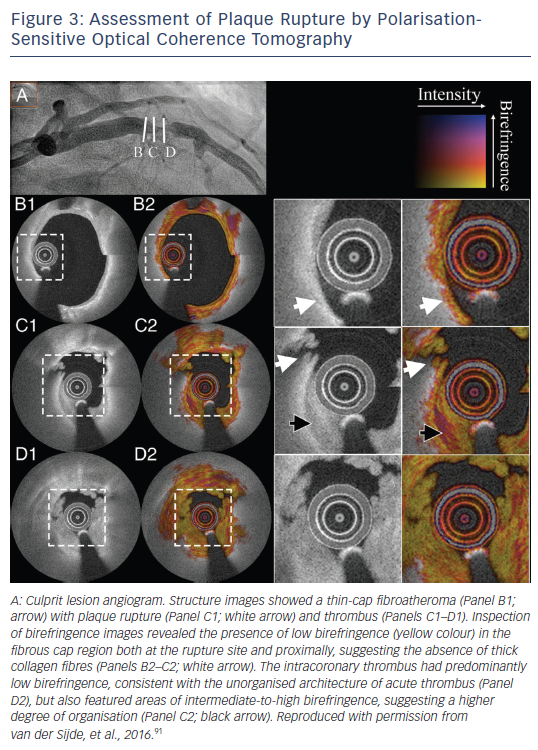
Technological advances include improved plaque characterisation using the current technology and the development of new technologies based on OCT imaging. Algorithms for the automated measurement of cap thickness are already available that can provide a fast and objective measure of this critical component of plaque vulnerability, while simultaneously increasing reproducibility of such measurements.85,86 Systems for automated tissue characterisation in atherosclerotic plaques can be used for a faster, more objective evaluation of plaque morphology.87,88 The development of ultra-high-speed OCT imaging systems will allow the complete high-resolution assessment of long arterial segments within a heartbeat,89 enabling a more accurate biomechanical assessment of the haemodynamic environment in the coronary arteries. The development of technologies such as polarisation-sensitive OCT that can measure the collagen content of atherosclerotic plaques90 will perhaps better redefine fibrous caps at a high risk of rupture (see Figure 3).91 Furthermore, new imaging systems with resolution comparable to electronic microscopy able to identify (sub-)cellular structures,92 and hybrid systems incorporating the ability for detection of molecules targeted with probes simultaneously with plaque imaging,93 are currently under development; these systems may enhance the prognostic potential of OCT imaging for vulnerable plaque identification.
Conclusion
Introduction of OCT in clinical practice has provided useful insights into the pathomechanisms of vulnerable plaque. These insights have enhanced our understanding of ACS, and have provided a knowledge base to use for clinical purposes. However, identification of the vulnerable plaque is not yet ready for clinical application, as the prognostic significance of a vulnerable plaque phenotype is poor, and a clinical benefit from interventions targeting vulnerable plaques is yet to be proven. Therefore, a better identification process needs to be implemented that can increase the prognostic significance of the findings in a way that it will be meaningful for the development and testing of local strategies targeting the vulnerable plaque. Future developments in the field will be aimed at early identification of highrisk plaques and timely intervention that could potentially result in a reduction of the mortality rates of patients with ACS.