










Non-alcoholic fatty liver disease (NAFLD) is characterised by the presence of steatosis in more than 5% of hepatocytes, often associated with metabolic risk factors (particularly obesity and type 2 diabetes), and in the absence of excessive alcohol consumption or other chronic liver diseases.1 NAFLD serves as an umbrella term encompassing a wide range of clinicopathological findings. From a histological perspective, NAFLD represents a disease spectrum that includes steatosis, either with or without mild inflammation (known as non-alcoholic fatty liver), and a necroinflammatory subtype (known as non-alcoholic steatohepatitis [NASH]). NASH is further characterised by the presence of hepatocellular injury, marked by hepatocyte ballooning.2
NAFLD is the most common chronic liver disease in developed countries, affecting approximately 25% of the world’s adult population.3 The natural course of the disease moves towards NASH and cirrhosis, implying that NAFLD will be the leading cause of liver transplantation in the coming years.4 Sedentary behaviours and poor eating habits, in parallel with the growth of metabolic diseases, such as obesity and diabetes, are the main causes of recent increases in the prevalence of NAFLD.5 Therefore, the clinical and economic burdens of NAFLD not only depend on liver-related mortality, but also extrahepatic diseases (e.g. type 2 diabetes) and an increased risk of cardiovascular disease (CVD).6
In this sense, Stepanova et al. reported that patients with metabolic syndrome (MetS) had a 40% increased risk of NASH, and a recent meta-analysis found that 71% of patients with NASH had MetS, 82% had obesity, 44% had diabetes and 72% had hyperlipidemia.7,8 Based on these findings, the accumulation of visceral adipose tissue (VAT) that usually accompanies NAFLD may be responsible, in part, for the comorbidities, with the increased release of free fatty acids into the circulation leading to the development of hepatic insulin resistance and hepatosteatosis. Indeed, the alterations in hepatic lipid metabolism that lead to NAFLD also drive the development of atherogenic dyslipidaemia, especially elevated plasma triglyceride (TG) concentrations, remnant lipoprotein cholesterol levels and small dense LDL (sdLDL) particles that infiltrate the arterial wall and promote the development of atherosclerotic plaques.9 Furthermore, altered glucose metabolism and insulin resistance, which are also hallmarks of NAFLD, can further exacerbate CVD risk in patients with NAFLD.9 Moreover, the increased release of inflammatory cytokines from VAT and the liver observed in patients with overweight contributes to the development of insulin resistance and NAFLD.10
Although there is considerable evidence linking NAFLD to CVD, there are still unresolved questions regarding this relationship. The most important question is whether the association between the two conditions is due to shared risk factors (obesity or diabetes) or whether hepatic steatosis itself increases the risk of atherosclerosis.11,12 In addition, it remains difficult to predict which patients with NAFLD will develop severe liver complications or CVD. Therefore, there is increasing interest in identifying patient characteristics that can help predict the progression of these diseases; closer monitoring and the development of new biomarkers of subclinical atherosclerosis are needed to detect individuals at high cardiometabolic risk who are candidates for therapeutic interventions aimed at preventing the progression of NAFLD and atherosclerotic CVD.13 This review briefly summarises some of the mechanisms responsible for the association between NAFLD and CVD, including novel concepts such as extracellular vesicles (EVs), proprotein convertase subtilisin/kexin type 9 (PCSK9), the microbiome and genetics factors.
Non-alcoholic Fatty Liver Disease and the Prevalence of Clinical Cardiovascular Disease
Several recent studies have reported that the risk of cardiovascular events (fatal and non-fatal) is increased in patients with NAFLD, with either simple steatosis or NASH, independent of other cardiovascular risk factors.14–17 This suggests that NAFLD could enhance the risk already present because of the underlying cardiovascular risk factors (e.g. hypertension, diabetes and dyslipidaemia).18 Moreover, the risk of fatal cardiovascular events is increased in patients with severe NAFLD, and these events are the most common causes of morbidity and mortality in patients with NAFLD compared with other liver-related causes (incidence of 4.79 versus 0.77 per 1,000 person/years).1,9 In addition, the annual incidence of atherosclerotic CVD in patients with severe NAFLD has been estimated at around 1.1%, which, taking into account the increasing prevalence of NAFLD, is expected to grow notably in the coming years.19
According to recent literature, NAFLD is an independent risk factor for the development of MI. Indeed, Sinn et al. showed that the increased risk of MI was evident in individuals with either a high or low NAFLD fibrosis score.20 A study of 2,103 Italian outpatients with type 2 diabetes reported a significant association between NAFLD and incident CVD, defined as MI, ischaemic stroke, coronary revascularisation or cardiovascular death.21 This association appeared to be independent of a broad spectrum of risk factors, thus suggesting that NAFLD may confer an excess CVD risk over and above what would be expected because of the increased prevalence of the underlying metabolic risk factors.21 In addition, in a prospective observational study of 1,637 Japanese individuals from a health check-up program, Hamaguchi et al. observed that NAFLD was a predictor of CVD (defined as coronary heart disease, ischaemic stroke and cerebral haemorrhage) independent of conventional risk factors.22
Subclinical atherosclerosis, as an early stage of CVD, has considerable clinical significance. We previously reported that NAFLD was associated with 2.04-fold higher prevalence of pathological carotid intima–media thickness (IMT) and 4.41-fold higher risk of carotid plaques.23 Similarly, a prospective study in 3,343 patients with NAFLD diagnosed by hepatic ultrasound concluded that increased brachial–ankle pulse wave velocity was independently associated with a higher risk of NAFLD and fibrosis, whereas pathological carotid IMT was associated with incident NAFLD but not fibrosis.24
Socioeconomic and epidemiological factors, such as ethnicity and geographic differences, can influence various facets of health linked to fatty liver disease and cardiometabolic factors.25 In this context, Juonala et al. found a link between neighbourhood-related issues in Australia and less severe adiposity in children with obesity.26 That study also found that access to shopping facilities (shops or other retail services) was connected to an increased risk of dyslipidaemia and fatty liver.26 Recently, a significant correlation was established between skill level (skill level 1 covers the most basic positions; skill level 2 requires the effective comprehension of information like safety protocols and basic arithmetic; skill level 3 demands advanced abilities and specialised knowledge; skill level 4 entails solving intricate problems and making decisions grounded in extensive theoretical and experimental expertise within a specific area) and fatty liver index within a large middle-aged population, although no significant association was observed between sex and the presence of fatty liver.27
Recently, an international group of experts suggested that NAFLD be redefined as metabolic dysfunction-associated steatotic liver disease (MASLD).28 The potential effect of this change in nomenclature on the prediction of CVD risk remains uncertain. In a novel meta-analysis, a greater number of CVD events were identified in MASLD compared with NAFLD, although the risk of CVD incidents associated with either definition did not differ significantly. These findings underscore the importance of clinicians maintaining a strong suspicion that individuals with MASLD may also present with concurrent CVD.15
Mechanisms Linking Non-alcoholic Fatty Liver Disease, Endothelial Dysfunction and Atherosclerosis
The most frequent phenotype of NAFLD is associated with metabolic dysfunction, manifesting as obesity and/or diabetes and some of the features of MetS.22 To date, several dysregulated pathways have been proposed as links between NAFLD and cardiometabolic syndrome, such as changes in lipid metabolism, insulin resistance, de novo lipogenesis, endothelial dysfunction and oxidative stress, together with host genetics and changes in the gut microbiota (Figure 1).29,30
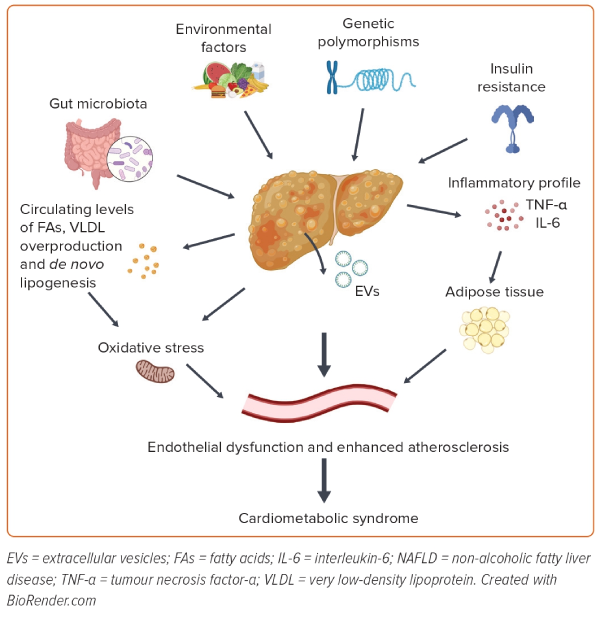
Lipid Dysregulation
NAFLD is frequently associated with dyslipidaemia, a proatherogenic condition characterised by elevated levels of TG-rich lipoproteins (TRLs), lower HDL levels and an increase in circulating sdLDL, which are more atherogenic than normal-sized LDL.31 The increase in circulating sdLDL is a CVD risk factor and has been observed frequently in patients with obesity and MetS.32 Comparing NAFLD and NASH patients, higher sdLDL levels are found in NASH patients, who have a higher risk of CVD.32 In addition, NAFLD is associated with changes in the HDL profile, particularly smaller HDL particles that have poor functionality and are cleared more quickly, contributing to lower HDL and apolipoprotein A1 levels.33 Finally, NAFLD is associated with prolonged postprandial lipaemia, which contributes to hepatic TG content and is an independent cardiometabolic risk factor.34 Elevated levels of TRLs, particularly those containing apolipoprotein B (ApoB), are a hallmark of NAFLD and represent a biochemical marker for lipoprotein alterations and CVD risk. The impaired clearance of ApoB–TRL particles by the liver is a key factor in the development of hyperlipidaemia and hepatic fat accumulation.35 This lipid profile may also be a consequence of decreased lipoprotein lipase (LPL) activity due to increased circulating levels of the inhibitors angiopoietin-like 3 (ANGPTL3) and angiopoietin-like 4 (ANGPTL4), which are increased in patients with NAFLD.35 In summary, dyslipidaemias play a key role in the development of NAFLD; therefore, the use of appropriate lipid-lowering therapy is crucial in the therapeutic algorithm of NAFLD to arrest the progression of liver disease and the associated CVD.36
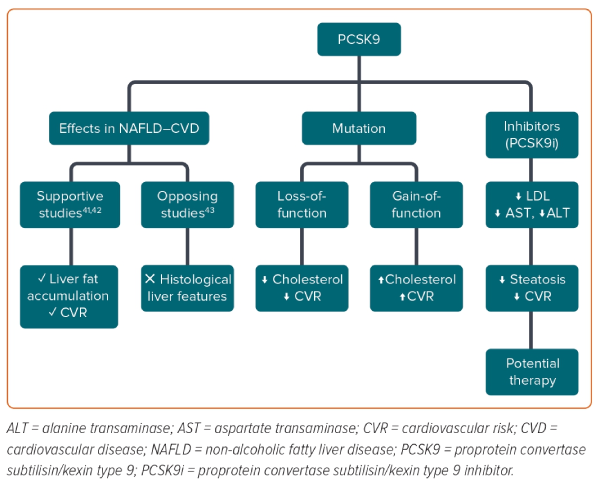
Proprotein convertase subtilisin/kexin type 9 (PCSK9), an enzyme mainly expressed in the liver, has emerged as a critical regulator of lipid homeostasis by acting as an inhibitor of LDL receptors, with a consequent increase in LDL levels and cardiovascular risk.37 In humans, PCSK9 loss-of-function mutations are associated with lower levels of cholesterol and protection against CVD.38,39 In contrast, a gain-of-function PCSK9 mutation was found to be associated with hypercholesterolaemia.38,39 Recently, PCSK9 has been linked to the severity of liver steatosis and the development of NASH, but the role of PCSK9 in NAFLD remains contentious.40 A recent study performed in NAFLD and obese patients after bariatric surgery found that the hepatic expression and circulating concentrations of PCSK9 were associated with the severity of steatosis and were inversely correlated with alanine transaminase (ALT) and aspartate transaminase (AST) levels; however, the study did not find a significant association with inflammation or ballooning.41 This supports, in agreement with other studies, the role of PCSK9 in liver fat accumulation and increased cardiovascular risk via increases in circulating cholesterol concentrations, but not in the progression of liver damage.42 Conversely, results from three cross-sectional studies from two hospitals in France with 132 patients did not find any association between circulating PCSK9 concentrations and histological liver features (Figure 2).43 Studies performed after the introduction of PCSK9 inhibitors could provide deeper insights. For example, Shafiq et al. reported a significant decrease in ALT and AST levels after 24 months of therapy with PCSK9 inhibitors in patients with NAFLD.44 In another study of 26 patients with familial hypercholesterolaemia, PCSK9 inhibitor therapy was found to significantly ameliorate steatosis in patients with low TG- HDL ratio (TG/HDL).45
Insulin Resistance and De Novo Lipogenesis
Dysregulation of glucose metabolism and insulin resistance are crucial common factors in NAFLD and in the pathogenesis of CVD.46 The presence of obesity or ectopic fat deposition and impaired insulin action in adipocytes promote a failure in lipolysis suppression, adipocyte stress and the recruitment and infiltration of macrophages, with the consequent release of proinflammatory cytokines, hormones and adipocytokines.47 The hyperinsulinaemia that accompanies insulin resistance promotes de novo lipogenesis and the maintenance of high glucose concentrations, which are responsible for the atherogenic dyslipidaemia that increases the risk of CVD.48 Insulin resistance also drives hepatic de novo lipogenesis, which contributes significantly to liver TG content, together with disturbed fatty acid oxidation (FAO) or altered secretion of very low-density lipoprotein (VLDL).49
Insulin activates transcription factors such as sterol regulatory element-binding protein 1c (SREBP-1c) and carbohydrate-responsive element-binding protein (ChREBP), regulating several enzymes involved in lipogenesis and pyruvate kinase, a master regulator of glycolysis.50 Indeed, diabetes is considered the most important atherosclerotic factor in NAFLD. Moreover, some glucose-lowering drugs, such as pioglitazone, have demonstrated promising benefits in ameliorating NAFLD.51 In addition, a recent meta-analysis showed that glucagon-like peptide 1 (GLP-1) and sodium–glucose cotransporter 2 inhibitors reduced the atherosclerotic risk.52
Thromboembolic Events
Targher et al. reported that the inflammatory profile associated with NASH may also play a pathogenic role in CVD.21 So, in the presence of systemic inflammation, the liver could act as a target of and a contributor to inflammatory changes. Hepatic steatosis is associated with the increased production of interleukin-6 and tumour necrosis factor-α by hepatocytes, Kupffer cells and stellate cells, and this contributes to the local activation of nuclear factor-κB.53 The biological link between NAFLD and CVD also includes prothrombotic factors. Venous thrombosis, a complex condition with multiple contributing factors, stands out as one of the most prevalent vascular diseases. The involvement of endothelial dysfunction plays a crucial role in the development of venous thrombosis.54,55 Acute and chronic liver diseases are associated with coagulation disorders.56 In this context, patients with NAFLD can show a prothrombotic state, and previous studies reported that NASH is an independent factor for the development of deep vein thrombosis and/or pulmonary embolism among patients with cirrhosis.57,58
Oxidative Stress and Endothelial Dysfunction
Oxidative stress is another factor involved in the pathogenesis of CVD, also in close relation to other chronic diseases, such as diabetes, MetS, obesity and NAFLD. In NAFLD, the excessive load of fatty acids in hepatocytes induces upregulation of the mitochondrial respiratory chain, resulting in overproduction of reactive oxygen species (ROS), exceeding the antioxidant capacity and leading to oxidative stress.52 The accumulation of ROS can activate Kupffer cells and stellate cells in the liver, leading to fibrosis and the progression of NAFLD. Indeed, the levels of circulating biomarkers of oxidative stress, such as urinary 8-iso-prostaglandin F2α and serum Nox2-derived peptide, are correlated with the severity of NASH.59
The accumulation of plasma oxidative stress contributes to the development of CVD by promoting endothelial dysfunction. The endothelium is a vital component of the vascular wall, and endothelial dysfunction, which is a hallmark of vascular diseases, is considered a marker of early atherosclerosis.60 Risk factors for atherosclerosis, such as hyperlipidaemia, hypertension, diabetes, smoking and infections, are common in patients with NAFLD. These factors can directly or indirectly stress the arterial endothelium, resulting in dysfunction or damage.61 Several pieces of evidence show that markers of endothelial dysfunction, such as flow-mediated dilatation percentage, are significantly impaired in patients with NAFLD, with a significant gradient according to the severity of liver disease.62 In addition, mean IMT, used as a non-invasive marker of endothelial dysfunction and arteriopathy, was significantly increased in patients with than without NAFLD.62 Patients with NAFLD had more carotid atherosclerosis than those without NAFLD, with enlarged mean IMT and higher plaque prevalence.63 This suggests that patients with NAFLD have an increased risk of CVD due to endothelial dysfunction.
Dysregulated Hepatokines
Emerging research has highlighted the role of dysregulated hepatokines in the pathogenesis of CVD in individuals with NAFLD.64 Fetuin-A and fibroblast growth factor 21 (FGF21) are implicated in insulin resistance, which is a major risk factor for both NAFLD and CVD.65,66 Conversely, FGF21 may reduce the risk of atherosclerosis by lowering inflammation, regulating lipid metabolism and adiponectin expression.67 ANGPTL3 can inhibit LPL activity.68 LPL activity encourages the healthy storage of TGs in the gluteofemoral compartment rather than in the pathological visceral compartment, thereby enhancing lipid metabolism and insulin sensitivity.69 Thus, elevated ANGPTL3 may contribute to cardiometabolic risk by inhibiting LPL. Finally, it has been shown that serum hepatocyte growth factor concentrations are elevated in patients with essential hypertension and extreme obesity, suggesting the role of hepatocyte growth factor in the pathophysiology of MetS and insulin resistance, and therefore in the increased cardiovascular risk.70
Genetics
Several pieces of evidence suggest that the NAFLD phenotype and differences in its progression are the result of complex interactions between the environment and an individual’s genetic pool. Recent studies have investigated the role of genetic polymorphisms associated with insulin signalling, type 2 diabetes, hypertension and lipid metabolism, all of which are shared risk factors for both NAFLD and CVD.71
The I148M variant of patatin-like phospholipase domain-containing protein 3 (PNPLA3) is the most known inherited determinant of NAFLD because it is associated with the progression of NAFLD, NASH and NAFLD-related hepatocellular carcinoma.72,73 This polymorphism induces a loss of function in enzyme activity, resulting in the accumulation of triacylglycerol in the liver and the entrapment of TG in lipid droplets in hepatocytes and hepatic stellate cells.69,74 This leads to accumulation of TG in liver cells, but a reduced secretion of VLDL into the circulation, which may reduce the deposition of lipids in the wall of blood vessels, reducing cardiovascular mortality.67 Supporting this scenario, extensive genomic research has linked the PNPLA3 I148M variant to a reduced risk of CVD.75
The transmembrane 6 superfamily member 2 (TM6SF2) E167K variant has been associated with higher TG levels in the liver and a greater risk of advanced fibrosis. It has also been reported to reduce ApoB lipidation, promoting a reduction in VLDL secretion from the liver; thus, controversially, patients harbouring this polymorphism showed a reduced cardiovascular risk.69,76 However, TM6SF2 upregulation may protect against cardiovascular events by reducing liver fat and increasing levels of circulating lipoproteins.77
Membrane-bound O-acyltransferase domain-containing (MBOAT7) is a gene implicated in the remodelling of phosphatidylinositol (and other phospholipids) via the incorporation of arachidonic acid and other unsaturated fatty acids into lysophospholipids. The common genetic variant leads to downregulation of MBOAT7 activity, and consequently to the accumulation of lysophosphatidylinositol in hepatocytes; this leads to a higher synthesis of TGs in the liver and NAFLD.69,78 Results regarding MBOAT7 and CVD are contentious; although Ismaiel and Dumitrascu reported no evidence of an association between MBOAT7 and cardiovascular risk, Xu et al. recently reported that MBOAT7 rs641738 (C>T) was associated with reduced metabolic traits and type 2 diabetes in elderly Chinese with NAFLD.79,80
In summary, several lines of evidence support that genetic variants associated with NAFLD are linked to a decreased risk of CVD or, at the very least, do not increase susceptibility to CVD. The observed connection between fatty liver and cardiovascular complications in epidemiological studies is likely primarily mediated by dyslipidaemia and insulin resistance, which are well-established risk factors for atherosclerosis. To definitively determine whether NAFLD genetics can impact CVD, we should take into account other genetic variations that predispose to NAFLD without affecting lipid secretion and plasma cholesterol and TG concentrations.81
Gut Microbiota
The gut microbiota is a complex ecosystem formed by different microorganisms, including bacteria, viruses, fungi and protozoa, that coexist in our gut through intricate relationships that also involve interactions with different organ systems of the host. The way in which these microorganisms behave, the metabolites they produce and the relative abundance of certain taxa have lately been related to health and disease.82 Disturbances to this symbiosis, known as dysbiosis, can alter the metabolic capacity of the system and impact the immune function of the host.83 Moreover, the gut microbiota can cause liver lipotoxicity and translocation of dysbiotic microorganisms or their products by increasing the permeability of the intestine barrier, inducing changes in the proinflammatory and anti-inflammatory balance in the liver, which has been demonstrated to be essential for the development of NASH.84 Indeed, disruption of the gut barrier seems to be a link between NAFLD and CVD because bacterial DNA was found in human atherosclerotic plaques.85 In addition, lipopolysaccharide, a component of the wall of Gram-negative bacteria whose levels are increased in NASH patients, can bind the endothelial surface, inducing an inflammatory response and endothelial dysfunction.86,87
Under normal conditions, up to 90% of the gut microbiota is formed by members of Bacteroidetes and Firmicutes, but an increase in the Firmicutes/Bacteroidetes ratio has been linked to CVD and NAFLD.88,89 The abundance of different taxa in the gut microbiota has been linked to CVD. On the one hand, Roseburia intestinalis and Faecalibacterium prausnitzii are both related to the amelioration of atherosclerosis, with R. intestinalis increasing fatty acid metabolism and reducing the inflammatory response in mice.90 On the other hand, Enterobacteriaceae, Ruminococcus gnavus and Eggerthella lenta are increased in individuals with atherosclerosis.91 Several taxa have also been shown to be related to NAFLD and its progression.92 However, technical and methodological challenges make comparisons between studies unreliable in most cases for analysing specific taxa. Bigger and better-designed studies in the future will allow a common nexus in the development of NAFLD and CVD to be found and, more importantly, how the functionality of the microbiota impact disease risk to be determined.
In this sense, metabolites produced by the gut microbiota can pass into the circulation, featuring systemic bioactive effects with inflammatory and metabolic properties. According to several studies, high circulating concentrations of trimethylamine N-oxide (TMAO) increase the risk of CVD, diabetes, NAFLD and other metabolic diseases.87,93,95 Trimethylamine (TMA), which is produced via metabolism of choline or l-carnitine by the gut microbiota, is oxidised to TMAO in the liver.95 TMAO has a proinflammatory effect, and studies have demonstrated that it is associated with cardiovascular events, NASH diagnosis and all-cause mortality.96 TMAO induces proatherogenic and prothrombotic mechanisms by promoting macrophage foam cell formation, decreasing liver cholesterol metabolism, modifying reverse cholesterol transport and causing endothelial dysfunction and platelet activation and aggregation.86 TMAO is related to endothelial cell dysfunction by elevating interleukin 6, C-reactive protein, tumour necrosis factor-α and ROS, while reducing nitric oxide production.97 Controversially, choline, from which TMA is produced, is essential for VLDL production and choline deficiency can lead to NAFLD by inducing hepatosteatosis and oxidative stress.98
Other microbially derived metabolites released by the microbiota after metabolising complex carbohydrates, such as short-chain fatty acids, can have an impact on the risk of CVD. These metabolites have been shown to improve lipid metabolism (decreasing LDL and TGs while increasing HDL), reduce inflammation and improve insulin sensitivity, all of which are risk factors for CVD and associated with NAFLD.99 In addition, by binding G protein-coupled receptor 41 and G protein-coupled receptor 43, these metabolites can activate the release of GLP-1 and peptide YY, regulating glucose homeostasis and appetite.100
Endothelium-derived Microvesicles as Biomarkers of Endothelial Dysfunction and Atherosclerosis in Patients With NAFLD
EVs, which are important mediators of intercellular communication, are lipid bilayer-delimited particles naturally released from almost all cell types that contribute to the pathogenesis, initiation and progression of several liver diseases and could be one of the mechanisms linking steatosis to the progression of atherosclerosis and endothelial dysfunction in patients with NAFLD.101 Growing evidence suggests that endothelium-derived microvesicles (EMVs; accounting for ~5–15% of all microvesicles) may be useful as novel biomarkers for CVD and metabolic diseases in adults, dyslipidaemia, endothelial dysfunction and MetS.102–105 In this context, Amabile et al. found that plasma EMV concentrations are associated with several cardiovascular risk factors, including higher TG concentrations, hypertension and MetS.103 We previously reported that plasma EMV concentrations decreased and endothelial dysfunction (measured by laser Doppler flowmetry) improved after liver disease improved in patients with chronic hepatitis C.106 In this context, EVs have been proposed as molecular mechanisms linking steatosis to the progression of atherosclerosis in patients with NAFLD. Jiang et al. isolated, quantified and characterised EVs from steatotic hepatocytes and demonstrated an inflammatory effect on endothelial cells via miR-1.107 These findings provide insights into the important role of EVs in the interaction between the liver and the vasculature. In agreement with the study of Jiang et al., Chen et al. isolated and characterised EVs from steatotic hepatocytes and concluded that these EVs promote foam cell formation and atherogenesis via the miR-30a-3p/ATP-binding cassette subfamily A member 1 transporter axis.108
Non-pharmacological Interventions and Their Potential Impact on Cardiometabolic Disease in Patients With NAFLD
A low-quality diet with elevated consumption of saturated fats and processed meat is a shared risk factor for the development of NAFLD, metabolic disorder and cardiovascular and all-cause mortality, confirming the complex interconnection between diet, metabolism, metabolic liver diseases and the cardiovascular system.109–111
In the absence of an approved effective drug for the treatment of NAFLD, the current first-line treatment for NAFLD consists of lifestyle modifications, including dietary, physical activity and weight-loss interventions.112 Following a calorie-restricted diet for a long period is linked to a reduction in cardiovascular risk and mobilisation of liver fat.113 However, only a low percentage of patients manage to stay on such a diet for a long period of time.114 Nevertheless, patients eating a Mediterranean diet showed improvements in steatosis independent of weight loss.115 In addition, another study found a reduced incidence of CVD in patients on a Mediterranean diet supplemented with either extra virgin olive oil or nuts compared with patients on a low-fat diet.116 Despite not achieving the goal of weight loss, a benefit on serum liver enzymes and the extension of steatosis have been reported with exercise alone.117 Most patients with NAFLD have a higher risk of MetS due to their lack of adherence to physical activity. Physical activity seems to be inversely associated with the prevalence of NAFLD, whereas BMI category is directly linked to NAFLD.118 Accordingly, a study in a Korean population showed a lower prevalence of or an improvement in NAFLD when patients performed moderate exercise five or more times per week.119 Furthermore, smoking should be avoided given that smoking is a risk factor for CVD and has been related to the progression of NAFLD.120 Thus far, merging diet and physical activity changes is the best advice for weight reduction in patients with NAFLD, also decreasing the risk of MetS.115 This approach boosts the reduction in CVD risk due to amelioration of the atherogenic risk and myocardial structure and function.121
Pharmacological Interventions and Their Potential Impact on Cardiometabolic Disease in Patients With Non-alcoholic Fatty Liver Disease
Several pharmaceutical drugs have been evaluated for the treatment of NAFLD. However, no single therapy has been approved thus far, although clinical trials are ongoing. Many of these clinical trials are based on peroxisome proliferator-activated receptor (PPAR) agonists.51,122–124 There are three types of PPAR nuclear receptors, namely PPARα, PPARδ and PPARγ, with PPARα being highly expressed in the liver.125 Activation of PPARα receptors stimulates FAO, thus protecting hepatocytes against oxidative stress-induced damage.125 In contrast, PPARγ is mainly located in the pancreas and in adipose tissue, with activation of PPARγ receptors promoting adipocyte differentiation, FAO levels in the liver and insulin sensitivity.125 PPARδ is ubiquitously expressed, and its function is to inhibit the oxidation of non-esterified fatty acids and lipogenesis. Moreover, PPARδ enhances anti-inflammatory outcomes.125 In a Phase 4 clinical trial, 58% of patients with NASH and diabetes achieved the primary outcome (reduction of at least 2 points in 2 histologic categories of the NAS without worsening of fibrosis after 18 months of therapy) after being treated pioglitazone, a first-generation PPARγ agonist (thiazolidinedione).51 Pioglitazone treatment improved hepatic steatosis, inflammation and swelling while decreasing TG concentrations and increasing HDL concentrations.51 The most important mechanism responsible for this improvement was the redistribution of lipids towards the subcutaneous adipose tissue.126 Pirfenidone, a PPARα agonist, inhibits adipogenesis and fibrosis.122 In the PROMETEO clinical trial (Phase 2), more than one-third of patients exhibited an improvement in fibrosis.123 Activation of PPARα has been related to improvements in atherosclerotic dyslipidaemia, inhibition of vascular inflammation and macrophage foam cell formation.121 This means that PPARα activation can prevent atherosclerotic progression and is why treatments for NAFLD also have effects on atherosclerotic cardiovascular disease (ASCVD).121
There are some treatments that include molecules acting on more than one receptor, and they are more potent. Lanifibranor, a pan-PPAR agonist, is able to activate PPARα, PPARδ and PPARγ. In a randomised control trial, lanifibranor treatment reduced steatohepatitis activity and improved fibrosis.124 Liver enzymes decreased and lipid biomarkers, inflammation and fibrosis were ameliorated.124 PPAR agonists also have benefits in the regulation of lipid metabolism, which means that not only can they be used for patients with NAFLD, but they can also be used to reduce ASCVD risk.121
Conclusion
NAFLD is a public health problem affecting up to one-third of the world’s adult population and is considered a multisystemic disease associated with extrahepatic alterations. NAFLD, especially in its more severe form (NASH), is associated with higher risk of CVD given the close association with cardiometabolic risk factors, with CVD being the leading cause of death among patients with NASH. There have been many studies investigating whether NAFLD contributes to cardiovascular risk, and it is plausible that treatment of liver disease may ameliorate the cardiovascular risk. Because there is no approved pharmacotherapy for NAFLD, these patients should be closely monitored and regularly screened for cardiometabolic risk factors. Treatment should focus on lifestyle interventions and lipid-lowering therapies, which will not only improve the liver, but also ameliorate the cardiovascular risk factors.
As described in this review, there are multiple factors in the aetiopathogenesis of cardiometabolic alterations associated with liver disease that need to be considered to determine the risk of cardiovascular disease in patients with NAFLD: genetic variants that serve as predictors of NAFLD are linked to a decreased risk of cardiovascular disease; TMAO, which is produced by the gut microbiota and oxidised in the liver, reduces nitric oxide production, enhancing endothelial dysfunction; and recent studies highlight the role of EVs in the progression of NAFLD-driven atherosclerosis. The development of precision medicine based on the development of biomarkers and therapeutic approaches considering proteomics, metabolomics, the microbiome and genetics (all of which are involved in the development of NAFLD and cardiovascular disease) would improve the management of patients with NAFLD. 