







Familial hypercholesterolaemia (FH) is the genetic disorder most commonly associated with elevated LDL cholesterol (LDL-C) levels from birth and with premature atherosclerotic cardiovascular disease (ASCVD).1 It is caused by mutations in genes related to the clearance of LDLs such as LDL receptor (LDLR), apolipoprotein B-100 (APOB) and proprotein convertase subtilisin/kexin type 9 (PCSK9).2 The prognosis for patients with FH has improved in the past 30 years, with statins improving clinical outcomes and reducing total mortality.3,4
Different strategies have been proposed to improve early detection of the disorder and ensure adequate treatment to prevent the development of ASCVD. The detection of index cases (IC; the first individual diagnosed in the family) through different case-finding strategies and cascade screening in relatives using LDL-C levels and/or genetic testing is feasible and cost-effective, especially for identifying cases in young people.5–7
This comprehensive review focuses on epidemiology, diagnosis and screening programmes, the goals of treatment and current lipid-lowering therapy in FH.
Prevalence
Traditionally, the prevalence of heterozygous FH has been estimated to be one case in 500 in the general population (0.2 %).2 However, a recent systematic review and meta-analysis has shown a prevalence of one in 250 individuals – higher than initially thought.8
Due to a founder effect, the prevalence of the disorder can be higher– up to one in 50–67 of the general population – in specific populations.9 In patients with genetically confirmed acute coronary syndromes, the prevalence of FH is up to 8.7 %.10,11
The prevalence of homozygous FH (HoFH) has been estimated to be one in 1 million, based on the frequency of heterozygous FH among relatives’ survivors of MI.12 However, a recent analysis established the prevalence of molecularly defined HoFH as being one in 300,000 individuals.13
Molecular Defects
FH is autosomal-dominantly inherited and >80 % of cases are caused by functional mutations in the LDLR gene. As of October 2017, >1,700 different functional mutations in the LDLR gene had been described worldwide.14,15 Five to 10 % of FH cases are caused by mutations in the APOB gene16,17 and <1 % are caused by some specific ‘gain-of-function’ mutations in the PCSK9 gene.14,18 These later mutations enhance the binding of PCSK9 to LDLR, increasing degradation of the receptor in the endosome, resulting in high cholesterol levels.19 There is emerging evidence that some patients with FH phenotype in whom a mutation in these three genes has not been detected may have a polygenic hypercholesterolaemia. These individuals have significantly higher number of LDL-C-increasing variations and LDL-C levels than controls.20
A very rare recessive form of FH that is clinically similar to HoFH is caused by mutations in low-density lipoprotein receptor adaptor protein 1 (LDLRAP1). The remaining FH cases are driven by monogenic mutations in other genes related to high cholesterol levels, such as APOE, SREBP2, STAP1 and LIPA.21
Cardiovascular Disease in Familial Hypercholesterolaemia
ASCVD in FH results from prolonged exposure to very high levels of LDL-C from birth.2 Vascular imaging studies in children have confirmed that carotid intima-media thickness is greater than in unaffected siblings by 8 years of age.22 Other studies using coronary angiography or electron beam tomography have shown that coronary atherosclerosis is evident from age 17 in males and 25 in females, and that 25 % of adolescents with FH have detectable coronary calcium.23,24
The first manifestation of the disorder is usually MI, which occurs as early as the third decade of life, and on average 20 years earlier than in individuals without FH.25,26 In the pre-statin era, a cumulative risk of fatal and non-fatal coronary events was observed in around 50 % of men and 30 % of women by the age of 60.27 The first UK Simon Broome Register analysis in 1991 showed that younger FH patients had a 100-fold increase in coronary mortality and nearly 10-fold increase in total mortality compared to the general normolipidaemic population.28 A recent analysis of the Copenhagen General Population Study showed a multifactorial adjusted odds ratio for coronary artery disease of 3.3 in carriers of a FH mutation.29 Data from the Spanish Familial Hypercholesterolemia Longitudinal Cohort Study (SAFEHEART) showed a prevalence of ASCVD in molecularly-defined FH subjects of 13 %, which is threefold higher than their unaffected relatives.30
Cardiovascular manifestations are highly variable depending on the molecular basis of the FH, LDL-C levels and the presence of other risk factors including lipoprotein(a).30–35 Patients with severe mutations are at higher risk than those patients with milder mutations.32,33 Lipoprotein(a) >1.8 µmol/l has been established as a cardiovascular risk factor in FH and its interaction with the type of mutation has been observed in molecularly-defined FH patients.34,35
HoFH is characterised by accelerated atherosclerosis that mainly affects the aortic root and coronary ostium. Absolute LDL-C levels are related to the severity of cardiovascular disease. The first cardiovascular event often occurs during early adolescence, especially in those cases with severe mutations (LDLR-negative). LDLR-defective patients usually develop clinical cardiovascular disease by the age of 30.36,37
Subclinical Atherosclerosis
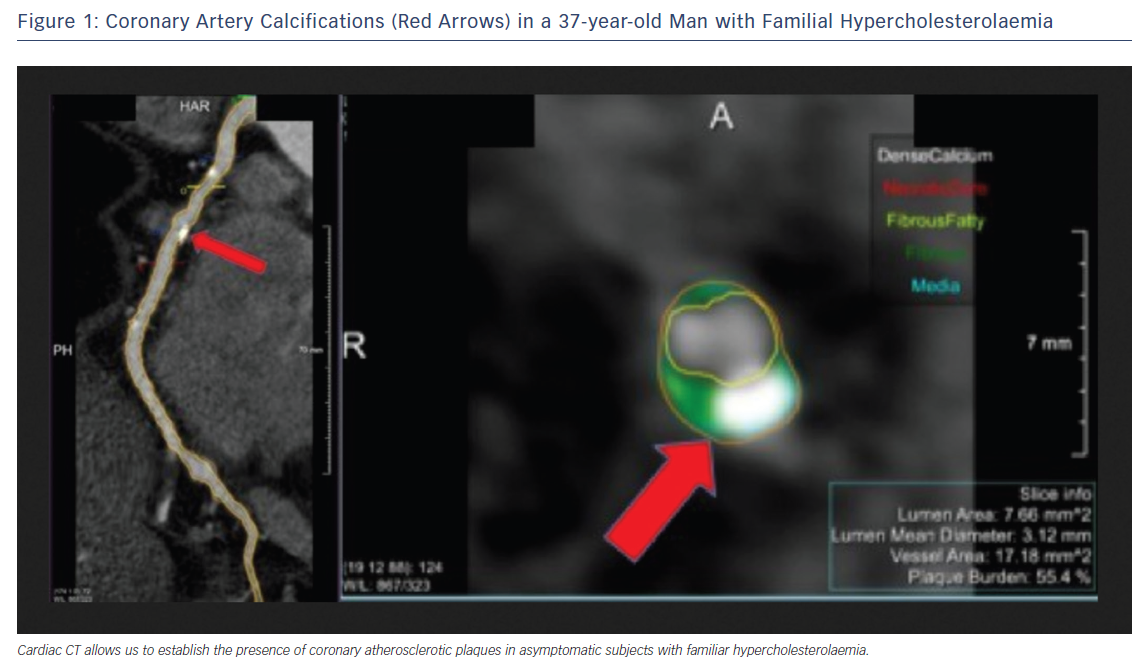
The atherosclerotic burden of FH can be demonstrated using non-invasive imaging techniques.38–40 In recent years, coronary CT angiography has emerged as a safe and non-invasive method of assessing coronary atherosclerosis.41 Cross-sectional studies have shown that FH patients have higher coronary artery calcium scores than non-FH individuals (Figure 1).42,43 However, it is still necessary to determine whether imaging studies improve risk stratification, intensity of treatment and clinical outcomes if they are incorporated as part of FH management.
Clinical Diagnosis of Heterozygous FH
A clinical diagnosis of FH is made based on high plasma levels of LDL-C, family history of hypercholesterolaemia, a history of premature ASCVD and the presence of tendon xanthomas.1,2 In general, LDL-C levels in adult patients are >4.9 mmol/l; however, lower cholesterol levels may be observed in some FH patients and their relatives, especially younger individuals, and an overlap in the distribution of LDL-C levels can be observed.44,45 Triglyceride levels are usually normal; however, high levels do not exclude the diagnosis if other criteria strongly suggest FH. Tendon xanthomas are pathognomonic for the disorder and are associated with higher cardiovascular risk.46,47 Xanthomas are present in <20 % of FH patients with a functional mutation;30,48 therefore, the absence of xanthomas does not exclude the diagnosis of FH.
In the past 30 years, three different clinical criteria have been developed for the diagnosis of FH. The MEDical PEDigrees with FH to Make Early Diagnosis and Prevent Early Deaths (MedPed) programme focuses on lipid levels, and does not incorporate clinical characteristics or genetic testing.49 The Simon Broome Register criteria for IC include lipid levels, tendon xanthomas, family history of hypercholesterolaemia, premature ASCVD and the presence of a functional mutation on genetic testing.6 Relatives should be diagnosed using gender- and age-specific LDL-C levels.50

The most accepted and commonly used FH diagnosis criteria are the Dutch Lipid Clinic Network criteria (Table 1). These criteria calculate a score based on LDL-C levels, the presence of arcus cornealis and tendon xanthomas, hypercholesterolaemia and premature CVD in relatives, and positive genetic testing. A total score ≥8 makes the diagnosis definite.51 These criteria should only be used for the identification of ICs.
Diagnosis in Children and Adolescents
FH should be suspected in children and adolescents where LDL-C >4.9 mmol/l after the exclusion of secondary causes of hypercholesterolaemia, or where LDL-C >3.9 mmol/l and one parent has confirmed FH.5–7,49 In the Netherlands, LDL-C >3.5 mmol/l can predict the presence of a LDLR mutation with a post-test probability of 0.98.52 Lipid levels vary with age, especially during puberty, and some overlap in LDL-C levels might be observed. Total cholesterol and LDL-C discriminate better among children with and without FH at 1–9 years of age.53 The affected parent should undergo genetic testing and, once the diagnosis has been confirmed, the implications of genetic testing in children should be discussed with them.5,54,55
Diagnosis of Homozygous Familial Hypercholesterolaemia
The diagnosis of HoFH is typically based on very high levels of LDL-C (untreated LDL-C >13 mmol/l or treated LDL-C >7.8 mmol/l on maximum lipid-lowering treatment) and the presence of cutaneous and tendon xanthomas in the first decade of life. Both parents must be heterozygous FH and should have elevated LDL-C levels.36 Phenotypic expression of this condition is highly variable depending on the type of mutation.37
Genetic Testing
Genetic testing is the gold standard for the diagnosis of the disorder and facilitates cascade screening. A pathogenic mutation in one of the LDLR, APOB and PCSK9 genes is identified in 70–80 % of definite FH cases and in 20–40 % in those with a milder phenotype.14,56 The Dutch Lipid Clinic Network criteria have better sensitivity and specificity than genetic testing.57 The absence of a known mutation does not exclude a diagnosis of FH, especially in those cases with a strong phenotype. Different studies have shown that using only cholesterol levels in ICs or relatives leads to the misdiagnosis of approximately 18 % of carriers and non-carriers of a mutation.58,59
In addition to the importance of confirming the diagnosis, a positive result from genetic testing is associated with prognosis. For any LDL-C level, individuals carrying a mutation are at higher risk for ASCVD than those who do not have a mutation.60 The type of mutation is also related to LDL-C levels and ASCVD risk, as shown in several studies.32,61
Screening Strategies
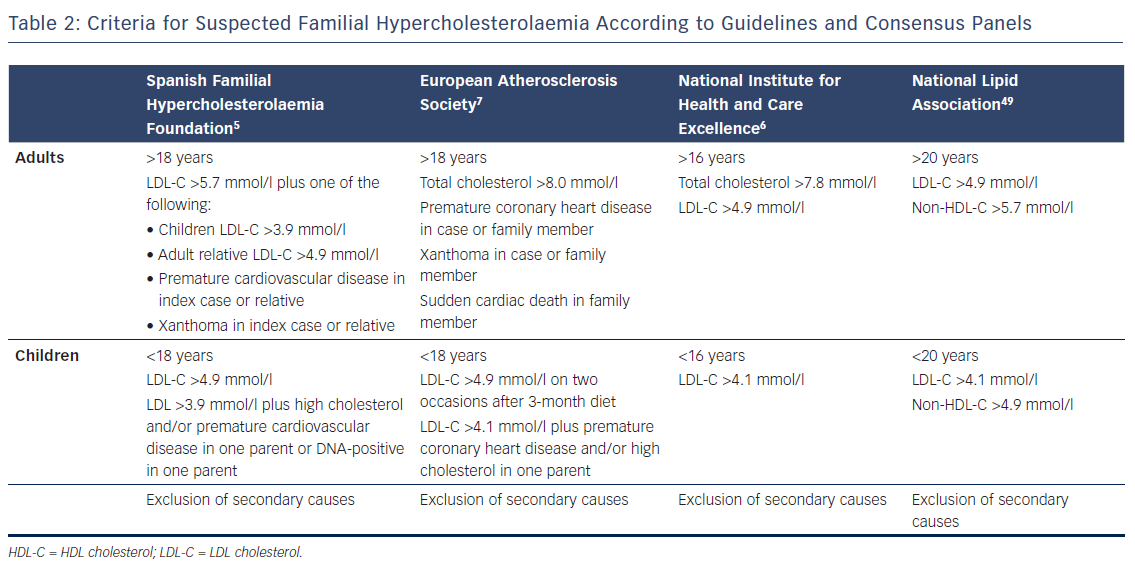
Early identification of FH is important for the prevention of coronary artery disease. At diagnosis, most patients are unaware of their condition and are receiving inadequate lipid-lowering therapy.62–65 ICs should be identified among individuals with total cholesterol >8 mmol/l, with cardiovascular disease <60 years of age and/or with tendon xanthomas or premature arcus cornealis (Table 2).6,7 When an IC is identified, the individual should be referred to a specialist for genetic testing, if available. Cascade screening using LDL-C measurement should be conducted in their relatives. If the mutation is known in the IC, consenting family members should also be offered a genetic test.6,7,49 Molecular testing avoids misclassification and is included as part of the screening algorithm in countries like Spain, the Netherlands and Norway.66,67 Different analyses have demonstrated that the identification of an adult IC through different case-finding strategies and cascade screening in relatives using lipid and/or genetic testing is the most efficient and cost-effective method of identifying new FH cases.64,68,69 Analysis of the SAFEHEART Registry predicted that identifying 9,000 cases of FH in 10 years could prevent 847 coronary events and 203 coronary deaths and add 767 quality-adjusted life years.69 Universal screening has the potential to detect more affected people in the community; for this, LDL-C levels should be measured in adults by the age of 20.49
Screening in children is still controversial, especially in relation to the age at which testing should be carried out and lipid-lowering therapy started.70,71 The US National Lipid Association recommends universal lipid screening of all children aged 9–11, and as early as 2 years of age if there is a family history of hypercholesterolaemia or premature ASCVD.49 In some countries, children are screened between 2 and 5 years of age as part of cascade screening in families with a known diagnosis of the disorder.5,53,55 In the UK, the National Institute for Health and Care Excellence guidelines recommend a genetic test is performed by the age of 10 in children who are at risk because they have one parent with FH.6
LDL-C Targets and Treatment of FH
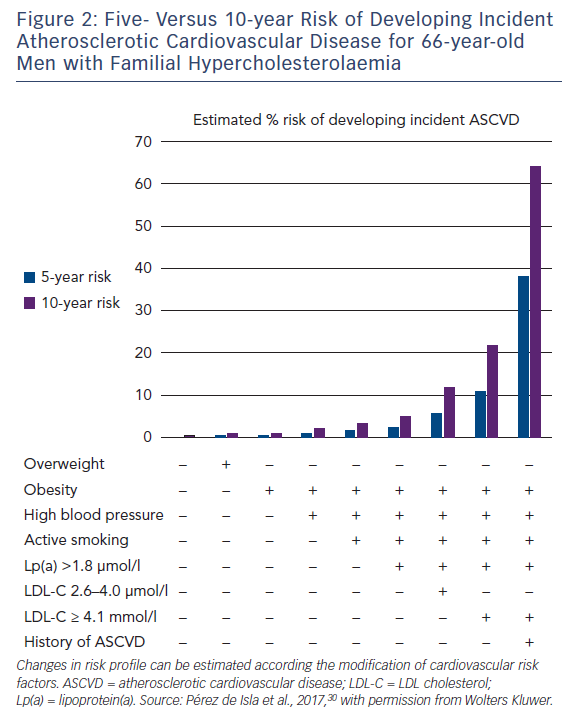
Observational data have shown that statins significantly reduce the risk of coronary and total mortality in FH.3,4,72 Intensive LDL-C reduction with statins or LDL apheresis also has beneficial effects on surrogate endpoints for ASCVD.73–75 Despite aggressive lipid-lowering therapy, FH patients still experience cardiovascular events, partly due to uncontrolled risk factors.76 Many studies have found that patients with FH are undertreated.77,78 A recent analysis of the SAFEHEART Registry showed that although most patients were on the maximum lipid-lowering therapy after 5 years of follow-up, only 11 % had reached the target of LDL-C <2.6 mmol/l.78 Despite the elevated lifetime risk for cardiovascular disease associated with FH, individuals’ risk profiles are heterogeneous. A cardiovascular risk equation including some classic risk factors and lipoprotein(a) and able to accurately predict incident ASCVD in molecularly-defined FH has recently been developed (Figure 2). Although recalibration with other populations is needed, this tool will improve risk stratification and treatment in FH patients.79
International guidelines consider LDL-C <2.6 mmol/l as the optimal target in adults with FH; <1.8 mmol/l in adults with FH and cardiovascular disease or type 2 diabetes; and at least a 50 % reduction in LDL-C levels if these goals cannot be achieved with maximally tolerated lipid-lowering therapy.5–7,49 Adults with FH should be treated from the moment of diagnosis.
There is no evidence to support a target LDL-C level in children. Expert consensus-recommended targets are <4.1 mmol/l,5,55 <3.5 mmol/l,5,70 <2.6 mmol/l80 and a reduction of 50 % from pre-treatment levels in children aged 8–10 years.70 The recommended age at which to start lipid-lowering therapy varies between 881 and 10 years,5,6,49,70,80 with5 or without70 differences between boys and girls. Analysis of 217 children <18 years old with FH in the SAFEHEART Registry found that the percentage achieving LDL-C <3.4 mmol/l increased from 20 % to 42 % after 4.7 years of follow-up. This result was principally explained by the use of statins.82
When pharmacological treatment is offered, the adult or child/adolescent or their parents or carer should be informed that treatment is lifelong.
Statins
All statins have been used in FH; however, most adult patients will require high-intensity statin therapy, such as atorvastatin 40–80 mg or rosuvastatin 20–40 mg.
In children with FH, treatment should be started at the lowest recommended dose and titrated up according to response and tolerability.5,70,82 In HoFH, statins can be started at age 2 and can produce a modest and variable effect on LDL-C, depending on the type of mutation.36
Women with FH should receive pre-pregnancy counselling. They should be given instructions to stop any lipid-lowering treatment at least 4 weeks before discontinuing contraception and should not use statins during pregnancy and lactation.
Ezetimibe
In patients with primary hypercholesterolaemia, ezetimibe monotherapy reduced LDL-C levels by about 18 %. It can be safely co-administered with statins, resulting in up to a 23 % incremental decrease in LDC-C in FH patients.83
Bile Acid Sequestrants
Bile acid sequestrants are rarely used due to their adverse gastrointestinal side-effects and poor patient compliance. Colesevelam has a greater potency to bind bile acids, providing a much better tolerability profile than the other sequestrants. Colesevelam reduces LDL-C by 13–19 % when administered as monotherapy and by an additional 18 % when prescribed in combination with statins.84
PCSK9 Inhibitors
Evidence that loss-of-function mutations in the PCSK9 gene produce low levels of LDL-C and lower the incidence of cardiovascular disease85 has further substantiated the role of PCSK9 as a potential target for the new generation of cholesterol-lowering drugs.
In 2015, the US Food and Drug Administration and European Medicines Agency approved evolocumab and alirocumab for the treatment of FH in patients who do not achieve LDL-C targets with maximum tolerated doses of conventional lipid-lowering therapy. The efficacy and safety of both these PCSK9 inhibitors have been demonstrated in FH patients with LCL-C inadequately controlled by statins and/or other lipid-lowering therapy.
A significant 50–60 % reduction in LDL-C over the reduction achieved by statins was obtained compared to placebo and they were well tolerated. Furthermore, >60 % of patients achieved LDL-C <1.8 mmol/l with PCSK9 inhibitors.86,87
The efficacy and tolerance of evolocumab have also been demonstrated in HoFH. A significant reduction in LDL-C levels of 20 % maintained after 48 weeks of treatment was observed in patients with and without apheresis.88 This reduction is modest compared with its effect in heterozygous FH. The reduced efficacy is because PCSK9 inhibition requires some LDLR activity, which is almost absent in HoFH.
Recent results from long-term outcome trials of PCSK9 inhibitors in patients with ASCVD have shown a significant 15 % relative risk reduction for major cardiovascular events, supporting their cardiovascular benefit.89,90 Although these trials were not carried out specifically in a FH cohort, they may also support the benefit of this class of drugs in this high-risk population.
Microsomal Triglyceride Transfer Protein Inhibitor
Lomitapide inhibits microsomal triglyceride transfer protein at the hepatocytes and enterocytes, preventing the assembly of triglycerides into very-low-density lipoprotein and chylomicrons. Lomitapide has been approved for the treatment of HoFH in people >18 years of age. LDL-C reductions of 50 % and 40 % at week 26 and 78, respectively, have been described.91 Due to its mechanism of action, gastrointestinal side-effects (mild transaminase elevations and diarrhoea) are the most common adverse events. These effects are managed by gradual titration of the dose and adherence to the recommended low-fat diet. Hepatic fat was found to be increased by up to 8 % at week 26 in patients taking lomitapide, but no further increase was reported for the 18-month duration of the study.91
LDL Apheresis
LDL apheresis is safe and is the only long-term treatment with the potential to slow early atherosclerosis and prolong survival in HoFH patients.92 LDL apheresis is also an option for patients with severe heterozygous FH, especially if pharmacological treatment insufficiently controls their LDL-C and their cardiovascular risk is still high.
Practical considerations have to be taken into account with this treatment. The cost, problems with insurance, venous access and the biweekly apheresis sessions are important issues that must be considered for each patient before proceeding.
Conclusion
FH is a common and treatable disorder. Early diagnosis and treatment will improve clinical cardiovascular outcomes. Identification of ICs and cascade screening using lipids and genetic testing in their relatives is cost-effective. Screening programmes are necessary to increase the number of cases identified and treated. Patients require high-intensity statin therapy and ezetimibe. For those not achieving target LDL-C, the new iPCSK9 are a good option for reducing LDL-C and cardiovascular risk. For severe HoFH, lomitapide and LDL apheresis are indicated.