










Platelet activation and aggregation play a pivotal role in the complex pathophysiological process of atherosclerotic disease, resulting in the formation of an occlusive thrombus.1 Furthermore, platelets modulate the various endothelial and inflammatory processes that underlie atherosclerosis. The amplification of platelet activation occurs through three main pathways: the cyclo-oxygenase 1 (COX-1) pathway, the ADP-P2Y12 pathway and the thrombin pathway. Antithrombotic therapies such as antiplatelet agents and anticoagulants work by targeting one of these pathways.1
Aspirin is the cornerstone of antiplatelet therapy, particularly in the secondary prevention of cardiovascular (CV) events. The active molecule in aspirin, acetylsalicylic acid (ASA), exerts its effect by inhibition of COX-1, which is necessary for the production of the platelet aggregation agonist thromboxane A2.2 Aspirin has been shown to lower the risk of major adverse cardiac events (MACEs) by up to 19% compared with placebo.3
Rivaroxaban is an anticoagulant that works by inhibiting factor Xa. As a key part of the platelet activation pathway, factor Xa is involved in the conversion of prothrombin to thrombin, a platelet agonist that facilitates fibrin formation.2
A dual-pathway inhibition (DPI) approach that combines an antiplatelet with an anticoagulant simultaneously blocks two of the pathways to thrombus formation for synergistic benefits.2
Major trials have demonstrated that the addition of rivaroxaban to aspirin is more effective in reducing CV risk than aspirin alone but may also increase the risk of bleeding. The ATLAS ACS-TIMI 46 and ATLAS ACS2-TIMI 51 trials showed that, in patients with recent (<7 days) acute coronary syndrome (ACS), the addition of rivaroxaban to aspirin reduced the risk of CV events and increased the risk of major bleeding and intracranial haemorrhage, but did not increase the risk of fatal bleeding.4,5 The COMPASS trial showed that this benefit was also apparent in patients with coronary artery disease (CAD) and peripheral artery disease (PAD).6 The VOYAGER-PAD trial found that, compared with aspirin alone, DPI therapy improved CV outcomes and increased International Society on Thrombosis and Haemostasis (ISTH) bleeding, but did not significantly increase thrombolysis in MI (TIMI) major bleeding in patients with PAD.7
Previous reviews and meta-analyses have investigated the benefit of DPI therapy over aspirin alone but have not excluded patients taking thienopyridines.8–12 Thienopyridines are irreversible ADP receptor/P2Y12 inhibitors that have an antiplatelet effect, and therefore reduce CV risk and increase bleeding risk. We performed a systematic literature review to identify studies comparing the effect of DPI therapy with aspirin alone in patients with CAD and PAD. We then performed a meta-analysis using data from these studies to compare the efficacy, safety and net clinical benefit (NCB) of DPI therapy with low-dose aspirin alone, excluding patients taking thienopyridine, to avoid possible confounding on both efficacy and safety.
Methods
This systematic review and meta-analysis were performed in accordance with Preferred Reporting Items for Systematic Review and Meta-analysis (PRISMA) guidelines.13
Search Strategy
We searched PubMed and Embase (https://www.embase.com) for publications reporting the clinical efficacy, safety and NCB of DPI (low-dose aspirin + low-dose rivaroxaban) compared with aspirin alone in patients with CAD and/or PAD in March 2022. Supplementary Tables 1 and 2 list the search strategies used for the two databases. Search parameters were limited to articles published in English after January 2017 to determine whether there were any trials following the publication of the COMPASS trial that would add to the findings on DPI therapy.6
Study Selection
The inclusion and exclusion criteria of publications were established prior to the conduct of the review and are detailed in Supplementary Table 3. A pilot screening was performed to ensure concordance between two screeners and reviewers with regards to publication inclusion eligibility. All citations were screened based on the title and abstract (first pass), followed by a full-text review (second pass) as per the eligibility criteria. Inclusion decisions were reviewed by a senior reviewer to resolve any discrepancies. In brief, this review included studies reporting the results of randomised controlled trials (RCTs; including any post hoc and subgroup analyses) that compared the clinical efficacy and/or safety of DPI (low-dose aspirin + low-dose rivaroxaban) with aspirin alone in patients with CV disease. Studies that did not report the outcomes of interest and those that focused on higher doses of aspirin >100 mg or rivaroxaban (>2.5 mg twice daily) were excluded. Efficacy outcomes of interest included MACE (defined as the composite of MI, stroke or CV death) and other outcomes, such as MI, any stroke, CV death and all-cause death. The safety outcomes were ISTH major bleeding and fatal bleeding events. NCB was measured as the composite outcome of CV death, stroke, MI, fatal bleeding events, and symptomatic bleeding into a critical organ.
Data Extraction and Quality of Evidence
The following were extracted from the studies and collated into a data extraction table: study characteristics (study name, study design, aim, study duration, country, inclusion and exclusion criteria, trial acronym, study groups and subgroups, and sample size); patient characteristics (age, sex, ethnicity, race, and geographical region); treatment characteristics (regimen and dose); baseline clinical characteristics (risk factors and coexisting conditions, history of atherosclerosis, PAD characteristics); and clinical outcomes reported for DPI versus low-dose aspirin including efficacy (MACE, MI, death), safety (stroke and bleeding), and NCB data. Internal validity was verified by a thorough quality check of the data.
Risk of Bias Assessment
Two reviewers (AK and RC) independently assessed the quality of each study using the Cochrane risk-of-bias 2.0 (RoB 2) Tool for randomised trials, a bias evaluation tool recommended by the Cochrane Collection.14 For each record, the RoB 2 tool assessed six domains: randomisation process (domain 1), deviations from intended interventions (domain 2), missing outcome data (domain 3), measurement of the outcome (domain 4), selection of the reported results (domain 5) and overall bias (domain 6). The risk of bias assessment satisfied the criteria of the A MeaSurement Tool to Assess systematic Reviews (AMSTAR) tool.15
Statistical Analysis
Data were analysed by two reviewers (AK and RC) using the meta package in R. A feasibility analysis was performed using Microsoft Excel. The feasibility analysis identified studies eligible for the meta-analysis based on the study characteristics, population characteristics, subgroups, treatment characteristics, HR as an estimate of treatment effect, 95% CIs and level of significance. The meta-analysis was performed with the individual and subgroup analyses split by population type and clinical outcome. The combined effect estimate was calculated using a logit transformation and inverse variance weighting.
Heterogeneity across the resulting data was evaluated using I2 statistics. Heterogeneity can be roughly interpreted as follows: 0–40%, might not be important; 30–60% may represent moderate heterogeneity; 50–90% may represent substantial heterogeneity; 75–100% considerable heterogeneity. A random effects model was used in the meta-analysis because there was some degree of heterogeneity in the study population and rivaroxaban dose across the studies. Pooled effect estimates that did not include the line of no effect were deemed statistically significant. A p-value <0.05 was considered to be statistically significant.
Results
Literature Search
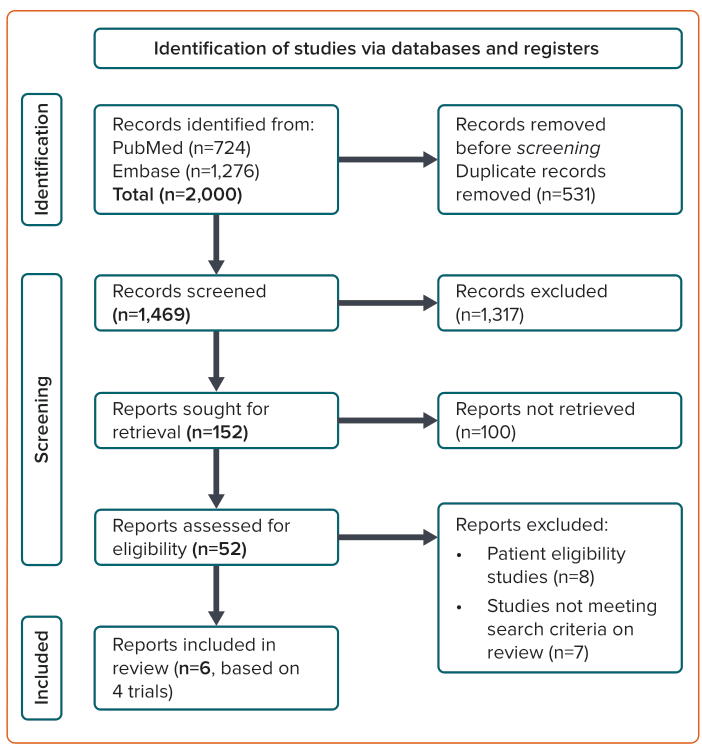
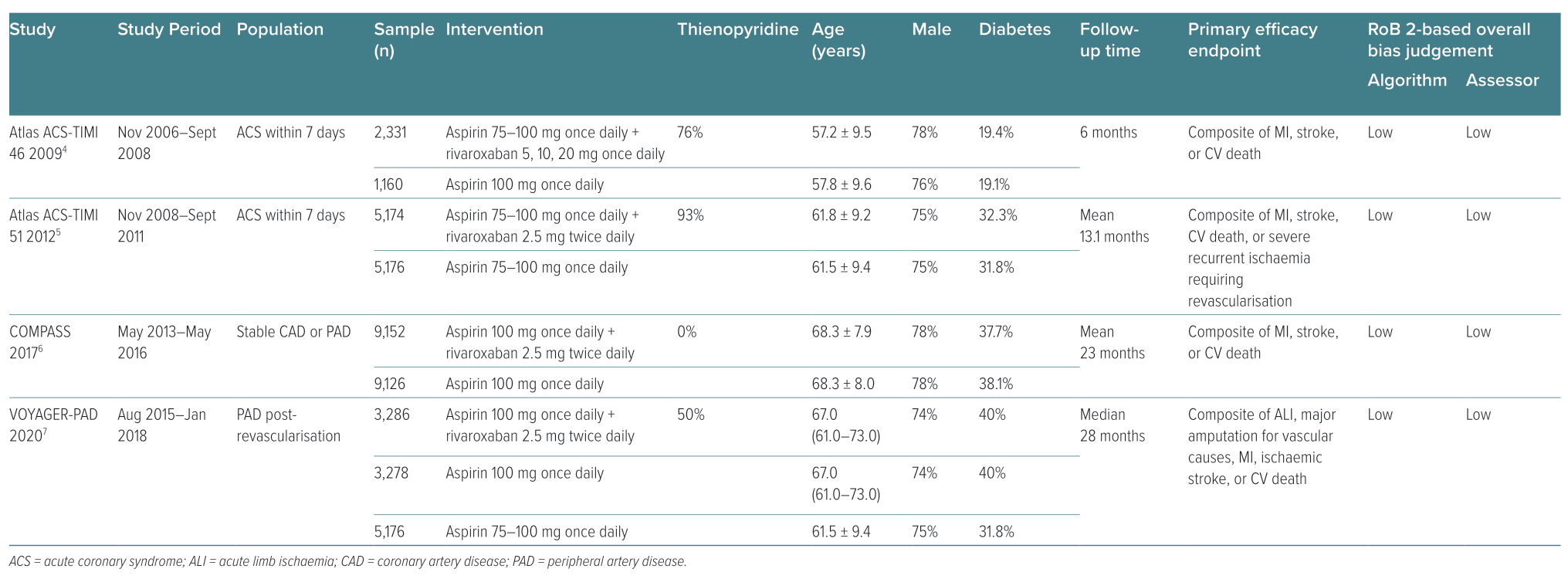
Of the 2,000 studies identified, 52 articles were assessed for eligibility and six articles representing four major trials were deemed suitable for inclusion in the meta-analysis (Table 1).4–7,16,17 One article was from the COMPASS trial, two were from the VOYAGER-PAD trial, one was from the ATLAS ACS-TIMI 46 trial, one was from the ATLAS ACS-TIMI 51 trial, and one was a pooled analysis of the ATLAS ACS-TIMI 46 and 51 trials (Figure 1).4–7,16,17 All six selected articles presented an overall low risk of bias based on the algorithm’s overall judgement and assessor’s overall judgement (Table 1). The COMPASS trial did not include any patients taking thienopyridines.6 In the VOYAGER-PAD trial, 50% of the patients were taking thienopyridines, but these patients were excluded in one arm of a subgroup analysis of the VOYAGER-PAD trial.7,16 A large number of patients in the ATLAS-ACS TIMI 46 trial (76%) and the ATLAS-ACS TIMI 51 trial (93%) were taking thienopyridines; however, a pooled analysis of these trials excluded patients taking thienopyridines.4,5,17 We considered only patients who were not taking thienopyridines for the purposes of this meta-analysis. The patient numbers for each group are listed in Supplementary Material Table 4.
Meta-analysis Results
Four RCTs included in the meta-analysis reported data for patients who had an ACS event in the last 7 days, stable CAD (mean time since an acute event: 7.1 years), and/or PAD. The median follow-up time varied among the four trials: ATLAS ACS-TIMI 46, 6 months; ATLAS ACS-TIMI 51, 13.1 months; COMPASS, 23 months; and VOYAGER-PAD, 28 months.4–7 Efficacy was measured using results from COMPASS and VOYAGER-PAD, as well as pooled ATLAS ACS-TIMI 46 and 51 data.4–7,17 Safety data were reported as ISTH major bleeding and fatal bleeding based on results from COMPASS, VOYAGER-PAD and ATLAS ACS-TIMI 51.5–7 NCB data were reported only in the COMPASS and VOYAGER-PAD trials.6,7
Efficacy
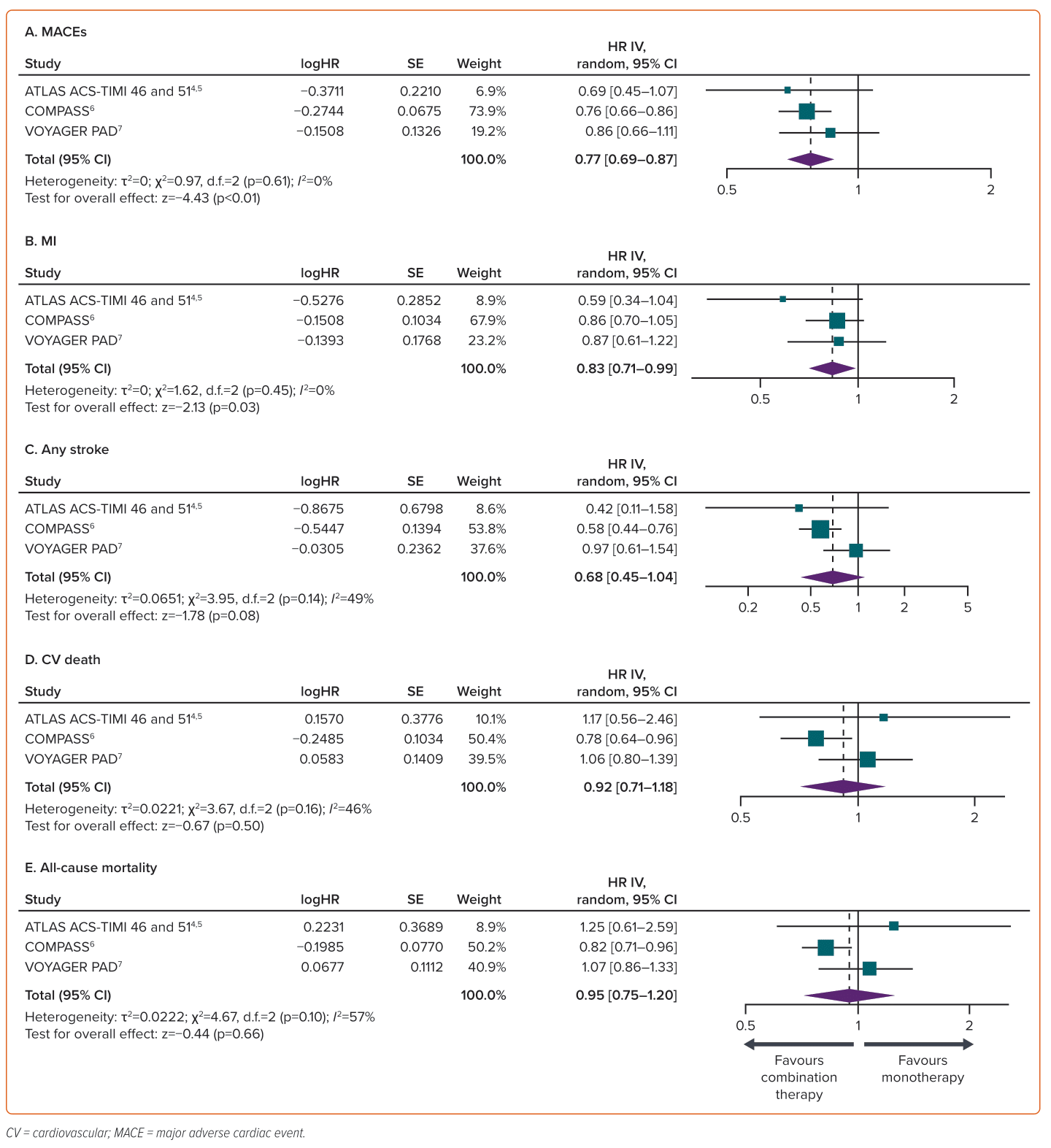
The incidence of MACEs, MI, stroke, CV death and all-cause death was reported in all four trials.4–7 There was a total of 714 MACEs (6.38%) among 11,199 patients in the group receiving aspirin and rivaroxaban, compared with 908 MACEs (8.00%) among 11,345 patients in the group receiving aspirin only. Adding low-dose rivaroxaban to aspirin compared with aspirin alone resulted in a significant reduction in the relative risk of MACE (23%; HR 0.77; 95% CI [0.69–0.87]; p<0.01) and MI (2.09% versus 2.68%; HR 0.83; 95% CI [0.71–0.99]; p=0.03). There was no remarkable decrease in the relative risk of stroke (1.13% versus 1.69%; HR 0.68; 95% CI [0.45–1.04]; p=0.08), CV death (2.31% versus 2.79%; HR 0.92; 95% CI [0.71–1.18]; p=0.50) or all-cause death (4.54% versus 4.97%; HR 0.95; 95% CI [0.75–1.20]; p=0.66) (Figure 2).
Safety
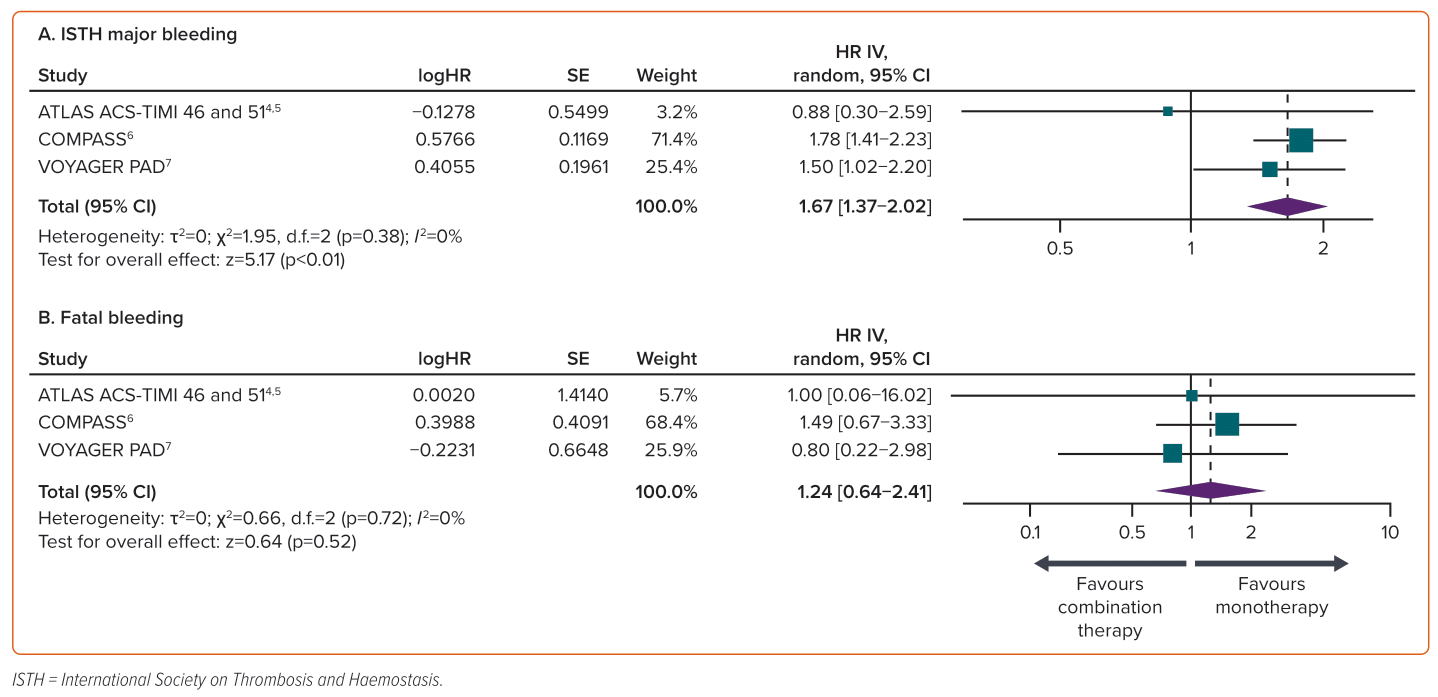
Four trials reported ISTH major bleeding and three reported fatal bleeding.4–7 The incidence of ISTH major bleeding events was significantly greater with DPI therapy compared with aspirin alone (1.75% versus 1.05%; HR 1.67; 95% CI [1.37–2.02]; p<0.01). The incidence of fatal bleeding did not differ significantly between DPI therapy and aspirin alone (0.12% versus 0.1%; HR 1.24; 95% CI [0.64–2.41]; p=0.52). In the ATLAS ACS-TIMI 51 trial, patients had a lower risk of ISTH major bleeding with DPI versus aspirin alone, and patients with PAD had a lower risk of fatal bleeding with DPI versus aspirin alone (Figure 3).5
Net Clinical Benefit
NCB, measured as the net clinical outcome of CV death, stroke, MI, fatal bleeding, or symptomatic bleeding into critical organ, was reported only in two studies.6,17 DPI therapy versus aspirin alone resulted in a favourable NCB. A significant reduction in total adverse events of 21% (4.84% versus 6.15%; HR 0.79; 95% CI [0.70–0.90]; p<0.01) was observed with DPI in patients in the immediate post-ACS phase and those with established CAD (Figure 4).

Discussion
The results of our meta-analysis show that, compared with aspirin alone, DPI therapy results in a significant reduction in the relative risk of MACEs, a significant increase in the risk of ISTH major bleeding, a similar incidence of fatal bleeds, and a significantly better NCB. Given that thienopyridines lower the risk of CV events and increase the risk of bleeding, we removed this source of bias from the analyses by excluding patients taking concomitant thienopyridines.
The ATLAS ACS-TIMI trials were the first to demonstrate the benefit of adding rivaroxaban to aspirin for the prevention of CV events; the COMPASS and VOYAGER-PAD trials have since demonstrated similar results.4–7
Combining the findings from these studies, we observed that DPI therapy resulted in an overall 23% reduction in MACE risk compared with aspirin alone, particularly in patients in the immediate post-ACS phase and patients with stable CAD and/or PAD. This effect was less prominent in patients with PAD alone. The greatest risk reductions in MI and all types of stroke were seen in patients in the immediate post-ACS phase. CV death risk was lower with DPI therapy only in patients with stable CAD; however, across the entire study population, there was an 8% reduction in CV death with DPI therapy versus aspirin alone. Similarly, although overall all-cause mortality was reduced by 5% with DPI therapy versus aspirin alone, a reduction in all-cause mortality was seen only in the COMPASS patient population.6 The overall trend of lower CV death and all-cause death may be driven largely by the fact that a large amount of the data used in this study came from the COMPASS trial.6
The safety of DPI therapy was assessed based on the incidence of ISTH major bleeding and fatal bleeding events. Although the overall risk of ISTH bleeding events was significantly higher with DPI than low-dose aspirin alone, it was not increased in patients in the immediate post-ACS phase (ATLAS-ACS TIMI 51 trial).5 Furthermore, the incidence of fatal bleeding events was lower in the VOYAGER-PAD trial and higher in the COMPASS trial with DPI therapy versus aspirin alone.6,7 There was no difference in the incidence of fatal bleeding events with DPI therapy versus aspirin alone in the ATLAS ACS-TIMI 51 trial.5
The NCB of DPI therapy was determined based on data from the COMPASS trial and the pooled analysis of the ATLAS ACS-TIMI 46 and 51 trials.4–6,17 DPI therapy resulted in an NCB of 21% relative to low-dose aspirin alone. In the VOYAGER-PAD trial, NCB was not reported in a way that allowed comparison with the other trial data.7 A subgroup analysis of the COMPASS trial data found that the addition of rivaroxaban to aspirin had the greatest effect on NCB in high-risk patient subgroups.18 It would be of value to determine whether a similar pattern is seen in ATLAS ACS-TIMI and VOYAGER-PAD data.4,5,7
The incidence of CV mortality and fatal bleeding was used to assess the efficacy and safety of DPI to provide a better perspective on the benefits and risks of using DPI therapy over aspirin alone. CV deaths were reported in all four trials, with 278 deaths (2.32%) recorded among 11,999 patients receiving DPI therapy and 317 (2.79%) among 11,345 patients receiving aspirin alone.4–7 Fatal bleeding events were reported only in three trials; 20 fatal bleeding events (0.12%) were recorded among 16,024 patients receiving DPI therapy, while 16 (0.10%) were recorded among 16,013 patients receiving aspirin alone.6,7,17
The use of DPI therapy is recommended by numerous international organisations in their guidelines. The European Society of Cardiology guidelines make a class 2a recommendation for the use of dual antithrombotic therapy for >12 months in patients at high risk of ischaemic events and without increased risk of major bleeding.19,20 These patients include those with diabetes that requires medication, a history of recurrent MI, any multivessel CAD, polyvascular disease (CAD + PAD), chronic kidney disease with estimated glomerular filtration rate 15–59 ml/min/1.73 m2, premature (<45 years) or accelerated (new lesion within a 2-year timeframe) CAD, or concomitant systemic inflammatory disease (e.g. HIV infection, systemic lupus erythematosus, chronic arthritis).
Applying these findings, along with guideline recommendations, enables a more personalised approach to the use of DPI therapy in the treatment of CV disease. While low-dose aspirin remains the cornerstone of therapy in the management of CV disease, these findings highlight that patients with a low bleeding risk may receive an additional clinical benefit from receiving DPI therapy over aspirin alone.
There are several limitations to this meta-analysis. First, the COMPASS trial (n=27,400 patients) was a much larger trial compared with the others included and hence contributed a disproportionate amount of data compared with the VOYAGER-PAD and ATLAS-ACS TIMI 46 and 51 trials.4–7 Second, the patient populations included in the meta-analysis differed by CV disease type and, therefore, also by level of disease risk and treatment requirements. Another important limitation is the lack of access to patient-level data: this meta-analysis was based upon study-level data only. Additionally, the safety parameters analysed were not the primary endpoints of all of the trials. Finally, although the time course of treatment varied across the trials included in this analysis, most trials had a relatively short follow-up period that precluded investigation of the long-term effects of DPI therapy.
Conclusion
This meta-analysis underscores the potential value of low-dose aspirin beyond its established use as a monotherapy. It highlights that some patients with CAD and PAD, including those in the immediate post-ACS phase and those with stable CAD, receive a greater benefit (reduced CV events) from DPI therapy than aspirin alone and, despite an increase in bleeding risk, derive an overall NCB. The personalisation of therapy through the careful selection of patients who may benefit from DPI therapy may reduce the burden of CV disease. 