







Pulmonary hypertension (PH) is a common finding in a wide range of diseases,1 some types (such as pulmonary arterial hypertension [PAH] of which are rare (see Table 1). PH is not only common, especially in patients with left heart disease and chronic obstructive pulmonary disease or interstitial lung disease, but is also associated with a dismal prognosis in these patients.1 If we consider that PAH is also related to a high morbidity and mortality risk, when left untreated, it is clear that early diagnosis and treatment of pulmonary hypertension is of paramount importance.
Diagnosis
According to the 2009 European Society of Cardiology (ESC)/European Respiratory Society (ERS) Guidelines on Pulmonary Hypertension,2 PH is defined as an increase in mean pulmonary arterial pressure (mPAP) ≥25 mmHg at rest as assessed by right heart catheterisation (RHC). This means that RHC remains the gold standard method for the diagnosis of PH. The same guidelines have suggested a five step diagnostic algorithm for identifying the presence, severity and cause of PH (see Figure 1).
First Step – Raising the Suspicion of Pulmonary Hypertension
Clinical presentation as well as risk factors for PH usually lead a physician to suspect PH and proceed to investigations. The clinical presentation is non-specific and may consist of breathlessness mainly on exertion, fatigue, chest pain, exertional presyncope or syncope, palpitations and cough. The fact that PH symptoms are non-specific is most likely the reason for the significantly delayed referral of PH patients to PH expert centres, and this may have serious consequences for their prognosis. The clinical examination in these patients may reveal an elevated jugular venous pressure, a right ventricular heave, a loud pulmonary component of the second heart sound, a pansystolic murmur due to tricuspid regurgitation (TR), hepatomegaly, ascites, peripheral oedema and/or cool extremities. Clinical presentation will also lead to classification of symptom severity into the World Health Organization functional class (WHO FC), which guides appropriate treatment. Risk factors for PAH are connective tissue disease, particularly scleroderma, HIV infection, portal hypertension, congenital heart disease, chronic haemolytic anaemia, use of drugs (anorexigens, amphetamines, cocaine, etc.) and family history of PAH. While for non-PAH the risk factors are mainly lung disease, cardiovascular risk factors for left heart disease (LHD) and history of venous thromboembolism (VTE).
The most appropriate screening test for PH remains transthoracic echocardiography (TTE). TTE can estimate the peak velocity of the TR jet (tricuspid regurgitation peak velocity [TRV]) and the right atrial pressure (RAP) based on the diameter of inferior vena cava and its respiratory variation, and thus pulmonary artery systolic pressure (PASP) by using the simplified Bernoulli equation (PASP=4 x TRV2 + RAP). TTE can also identify additional echocardiographic variables suggestive of PH such as right ventricular (RV) dilatation, hypertrophy and/or systolic impairment of RV, interventricular septal (IVS) deviation, right atrial dilatation, RV free wall S wave on tissue Doppler imaging (TDI) <11.5 cm/s,3 tricuspid annulus plane systolic excursion (TAPSE) on M-mode <20 mm,4 pulmonary flow acceleration time <93 ms,5 RV isovolumic relaxation time >65 ms6 and RV myocardial performance index – Tei index >0.36.7 In addition, TTE can detect left ventricular (LV) systolic and diastolic dysfunction, valvular disease, and congenital defects which all can be potential causes of PH. The 2009 ESC/ERS Guidelines have proposed some arbitrary criteria according to the above-mentioned echocardiographic measurements and variables to classify PH as likely, possible or unlikely. According to these criteria a patient with TRV ≥2.8 ms can possibly have PH, especially in the presence of additional echocardiographic variables suggestive of PH. If PH is possible the physician should refer the patient to a PH expert centre for further investigations.

Second Step – Excluding Left Heart Disease and Lung Disease
The second step of this diagnostic algorithm is to diagnose the most common causes of PH that is LHD and lung disease. History and TTE are the most useful tools available to establish LHD. The presence of cardiovascular risk factors for LV systolic and/or diastolic dysfunction such as systemic hypertension, diabetes mellitus, coronary artery disease (CAD), obesity, atrial fibrillation (AF) and valvular disease raise the suspicion of LHD. TTE can offer further evidence for its presence. Echocardiographic findings that can cause LV diastolic dysfunction (LVDD) are: LV systolic dysfunction (which is almost always accompanied by LVDD); LV hypertrophy (LVH); the presence of at least moderate aortic or mitral valve disease; LV outflow tract obstruction (hypertrophic cardiomyopathy, subaortic stenosis etc.); restrictive cardiomyopathy; or constrictive pericarditis.
The echocardiographic diagnosis of LVDD needs to be based on the measurements described below.
- E/A (early [E] filling velocity/late [atrial-A] filling velocity) ratio measured from the pulse wave Doppler of mitral inflow.
- E/E’ (early [E] filling velocity on transmitral Doppler/early [E’] relaxation velocity on tissue Doppler) based on Tissue Doppler Imaging E’ measurement at the base of septal or lateral LV wall. E/E’
- >15 indicates increased LV filling pressures, while E/E’ <8 indicates normal diastolic function.
- Left atrial volume indexed (LAVi) by body surface area measured by the biplane Simpson’s method in four- and two-chamber views. LAVi >40 ml/m2 is considered to be a marker of severe LVDD.
- Pulse wave Doppler of the pulmonary veins flow. A diastolic wave bigger than the systolic and pulmonary vein flow duration greater than the mitral flow duration at atrial contraction usually indicate at least grade II LVDD.8,9
Unfortunately, the echocardiographic measurements of LVDD have some limitations, which are listed below.
- E and A waves can be fused when the patient is tachycardic or in the presence of frequent ectopic beats, making the interpretation of E/A ratio difficult.
- E/E’ is frequently in the grey zone (8–15). In addition, in constrictive pericarditis E/E’ is not indicative of LV filling pressures as e’ is preserved or increased despite the presence of LVDD. Measurement of E’ is generally thought to be more reliable at the septal rather than the lateral wall but in patients with PH, who often present with significant IVS deviation on the echocardiogram, septal E’ is low not because of intrinsic LVDD but because of the ventricular interdependence and the overloaded right ventricle. In these cases it is preferable to measure E’ at the lateral wall.10
- In the presence of AF the diagnosis of LVDD based on mitral inflow is not feasible because of the absence of the A wave, while increased LAVi can be misleading.
- In the presence of at least moderate mitral valve disease (stenosis or regurgitation) E/A ratio may not be reliable for diagnosis of LVDD as E wave is elevated because of the increased forward blood flow through the valve and not necessarily because of increased LV filling pressures.
- Finally, Doppler of pulmonary venous flow is obtainable in approximately 70 % of the patients.
At the same time IVS deviation caused by right ventricular dilatation and pressure on the LV can be a cause of increased LV filling pressures even if there is no intrinsic LVDD. A recent study in patients with idiopathic and heritable PAH showed that LVDD diagnosed by echocardiogram was prevalent in 90 % of these patients and grade I was by far the most frequent grade of LVDD (88 %).11 This study shows that a patient with PH and grade II or more of LVDD on echocardiogram has PAH. In the case that the echocardiographic diagnosis of LVDD is ambiguous right and left catheterisation is the gold standard method to directly measure LV filling pressures by measuring pulmonary capillary wedge pressure (PCWP) and/or LV end-diastolic pressure (LVEDP). Haemodynamic measurements can also have some limitations. One of these limitations is that these haemodynamic parameters are measured at rest and may lead to underdiagnosis of LVDD in patients who have normal LV filing pressures at rest and markedly increased on exercise. Furthermore, patients who undergo catheterisation are often ‘optimally’ diuresed and in this case LV filling pressures may be misleadingly low. Exercise or fluid challenge during catheterisation have been proposed in order to identify occult LV dysfunction but these tools do not have a defined normal range.
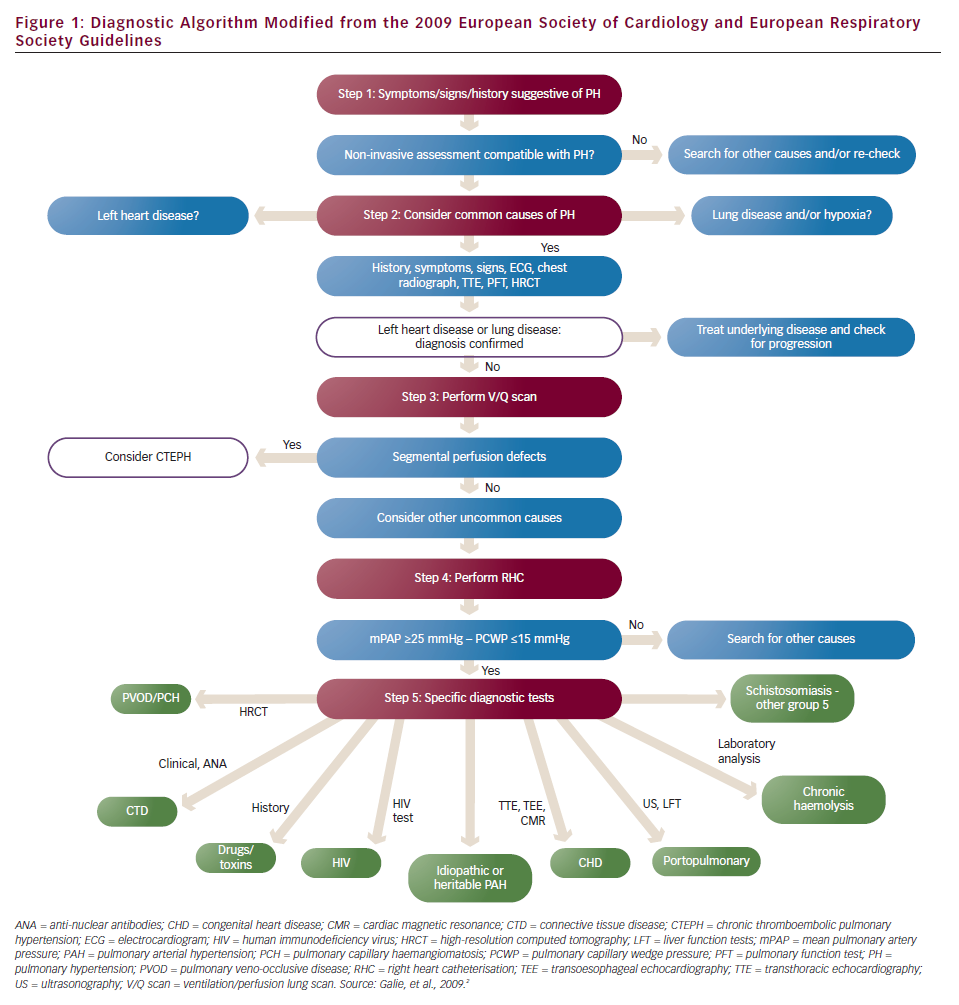
Establishing a diagnosis of lung disease requires lung function tests to measure spirometry, lung volumes and lung diffusion capacity for carbon monoxide, a high resolution computed tomography (CT) scan to identify emphysema or interstitial lung disease and arterial blood gases. Obstructive sleep apnoea syndrome can also be a serious cause of hypoxia associated with PH and should be excluded either by overnight oximetry at a minimum or by a polysomnography study. Additional investigations at this stage should include an electrocardiogram (ECG), a chest X-ray and routine blood tests including full blood count, renal, liver, thyroid function and brain natriuretic peptide (BNP). ECG can give valuable information about the LV such as the presence of LVH, ischaemia or previous myocardial infarction and left bundle branch block, while the presence of right ventricular strain has been related to PAH in previous studies.12 According to the diagnostic algorithm, when the diagnosis of LHD or lung disease is established and the severity of PH is not considered to be ‘out of proportion’ to the underlying disease, then there is no need for further investigations. Unfortunately, there is no adequate evidence at the moment to clearly define ‘out of proportion’ PH to the underlying LHD or lung disease and this term should be abandoned.
Third Step – Excluding Chronic Thromboembolic Pulmonary Hypertension
At this stage, chronic thromboembolic pulmonary hypertension (CTEPH), a rare but potentially treatable cause of PH should be excluded. A history of VTE is important as 74.8 % of CTEPH patients mention a history of acute pulmonary embolism.13 The screening method of choice to detect perfusion defects in the lungs is the ventilation/perfusion scan (V/Q scan), which is more sensitive than contrast CT angiography of the pulmonary artery (CTPA). If the V/Q scan is normal or of low probability then CTEPH can be ruled out with a sensitivity of 90–100 % and a specificity of 94–100 %.14 If the V/Q scan is of intermediate or high probability another method such as CTPA or magnetic resonance angiography (MRA) will be needed. These methods can establish the diagnosis and also identify the location of the thrombi n the pulmonary arteries, which is crucial for the patients’ eligibility for a pulmonary endarterectomy (PEA). Finally, selective pulmonary angiography remains the gold standard method for detecting filling defects in the pulmonary tree but, as it is an invasive method and requires a high level of expertise, should be used when other methods are not conclusive or PEA is considered.
When CTEPH is diagnosed RHC with or without a coronary angiogram (assessment of coronary arteries is usually essential in patients >40 year old or with history and/or risk factors for CAD) is necessary as pulmonary vascular resistance (PVR) is one of the important factors determining suitability for surgery and the outcome of PEA.15 RHC is also the next step of the algorithm to diagnose PAH, when CTEPH has been ruled out.
Fourth Step – Right Heart Catheterisation Study
RHC is useful in diagnosing PH, assessing its severity and testing vasoreactivity. If mPAP is <25 mmHg the diagnosis of PH is excluded and another cause to explain patients’ symptoms should be sought. At the same time, mPAP ≥25 mmHg measuring PCWP helps to classify the haemodynamics as pre- or post-capillary. When PCWP is ≤15 mmHg haemodynamics are characterised as pre-capillary and when it is >15 mmHg as post-capillary.2 Having excluded lung disease and CTEPH the presence of pre-capillary haemodynamics establish the diagnosis of PAH and further investigations to find an identifiable cause is needed. When RHC shows post-capillary haemodynamics, the diagnosis of PH associated with LHD should be reconsidered.
A vasoreactivity test is useful for the treatment of PAH patients, as true responders are likely to have a long-term response to high doses of calcium channel blockers (CCB). The most commonly used agent to assess vasoreactivity is inhaled nitric oxide (NO), which is short-acting with minimal systemic effects. The test is considered to be positive when a reduction of mPAP ≥10 mmHg to reach an absolute value of ≤40 mmHg with an increased or unchanged cardiac output is found. Vasoreactivity is uncommon (<10 % of PAH patients) and may be lost at a later stage.
Fifth Step – Identifying Causes of Pulmonary Arterial Hypertension
If the cause associated with PAH is not clear from the patients’ history, further tests need to be done. Immunology and serological tests are necessary to exclude connective tissue disease, HIV infection or hepatitis. An abdominal ultrasound is useful to diagnose portal hypertension and a TTE-bubble study or a transoesophageal echochardiogram can detect intracardiac shunting. If no cause can be found then the diagnosis of idiopathic PAH (IPAH) is made.
Two rare conditions with a poor prognosis associated with PH are pulmonary veno-occlusive disease (PVOD) and pulmonary capillary haemangiomatosis (PCH). The definite diagnosis of both conditions can be established by lung biopsy but this is a high-risk procedure and should not be attempted in PH. Hence, the diagnosis in most of the cases is based on the findings of high-resolution CT scans, which are characteristic but not pathognomonic. These findings include centrilobular ground-glass opacities, diffuse thickening of the interlobular septa and mediastinal lymph node enlargement.15 In this case, pulmonary oedema and LHD, especially in the presence of pleural effusions, should always be part of the differential diagnosis. Other tests such as bronchoalveolar lavage (BAL) are not well established for the diagnosis of PVOD/PCH.
Treatment
Evidence for treatment of PH with specific disease targeted drugs exists only for PAH. PAH-targeted treatments include three different classes of drugs that act on three different pathophysiological pathways:
- endothelin receptor antagonists (ERA) which target the endothelin pathway and include bosentan and ambrisentan;
- phosphodiesterase type-5 inhibitors (PDE-5i) which target the NO pathway and consist of sildenafil and tadalafil; and
- prostacyclin analogues which target the prostacyclin pathway and include epoprostenol, iloprost and treprostinil.
Non-pulmonary Arterial Hypertension Treatment
When PH associated with LHD or lung disease has been diagnosed, PAH-targeted vasodilatory treatments should be avoided since neither safety nor efficacy have been established. Trials with intravenous (IV) epoprostenol in systolic heart failure showed a higher mortality in the active treatment group,16 while large trials with ERA in LHD failed to show any benefit.17,18 Sildenafil has shown encouraging results in small studies with patients with LHD19,20 and is currently being tested in PH patients with normal LV systolic but LVDD, in the ongoing RELAX (Evaluating the Effectiveness of Sildenafil at Improving Health Outcomes and Exercise Ability in People With Diastolic Heart Failure) trial.21 Thus, in patients with cardiovascular risk factors for LV systolic and/or diastolic dysfunction or with valvular disease, treating the underlying disease and optimally controlling the risk factors is the recommended therapy at the moment.
In PH patients with lung disease there is not sufficient evidence for treatment with PAH-specific medications, as only short-term studies have been performed. In these patients treating the underlying lung disease is the only approved treatment at present. In addition PAH-specific therapy risks worsening ventilation/perfusion matching.
In CTEPH patients’ lifelong anticoagulation with a target international normalised ratio (INR) between two and three is the mainstay of treatment. Some of these patients are also eligible for PEA. PEA is a surgical procedure during which the surgeon physically removes accessible thrombi from within the wall of the pulmonary arteries. This operation requires a high level of expertise, must be done only in high volume centres and is suitable for CTEPH patients with mostly proximal organised thrombi in the pulmonary arteries, pre-operative PVR of <1,000 dynes/s/cm-5 and absence of significant co-morbidities.22 In inoperable cases there is lack of strong evidence for treating with PAH-specific therapies.
PAH has no curative treatment. PAH-targeted treatments aim to delay clinical deterioration, stabilise symptoms, increase exercise capacity and improve survival. These aims can be reached if a ‘treat-to-target’ rationale is followed. That means that therapy should focus on achieving specific targets such as WHO FC at least II, a six-minute walk test distance (6MWTD) more than 380 m (but higher in young patients), a peak oxygen consumption on cardiopulmonary exercise test more than 10.4 ml/min/kg and a peak systolic blood pressure >120 mmHg during exercise. The three above-mentioned drug classes are the appropriate treatment for PAH (see Figure 2).
Endothelin Receptor Antagonists
ERA inhibit the receptors of endothelin, a potent endogenous vasoconstrictor. Bosentan, an oral non-selective ERA, was evaluated in PAH and in patients with Eisenmenger syndrome as well as in PAH patients in WHO FC II and showed an increase in 6MWTD, improvement of WHO FC and haemodynamic status.23–25 Ambrisentan, an oral selective ERA, showed similar results in three trials.26–28 The major side effect of ERAs based on the results of randomised controlled trials23–28 is the increase of hepatic aminotransferases in approximately 10 % of the patients with bosentan and 2 % with ambrisentan. Less serious adverse effects of bosentan are anaemia and peripheral oedema. Ambrisentan is administered once daily, while bosentan twice daily. Both drugs require monthly checking of liver function tests.
Phosphodiesterase Type-5 Inhibitors
PDE-5i inhibit the enzyme which degrade cyclic guanosine monophosphate (cGMP), a potent vascular vasodilatory agent. Sildenafil and tadalafil in randomised clinical trials show amelioration of exercise capacity and haemodynamics as well as prolongation of time to clinical worsening in PAH patients.29,30 Sildenafil needs to be administered three times daily and tadalafil once daily. Their main side effects include headaches, flushing, epistaxis, indigestion, diarrhoea and muscle pain/cramps. In addition, these drugs should not be used concomitantly with nitrates because of the high risk of profound hypotension.
According to the 2009 ESC/ERS Guidelines ERA and PDE-5i are the recommended medications for PAH patients in WHO FC II and III. As the only head-to-head trial which compared the two drug classes did not show any difference in efficacy and safety, PH physicians can use any of them as their first choice.31 When the patient is in WHO FC IV or the first-line treatment does not achieve its targets, then prostacyclin analogues become the most appropriate option.
Prostacyclin Analogues
Prostacyclin analogues replace endogenous prostacyclin, a potent vasodilator, which is diminished in the pulmonary arteries of PAH patients. There are three approved agents in Europe: epoprostenol, treprostinil and iloprost. Epoprostenol has a very short half-life (approximately 1–2 min) and is unstable at room temperature. That is the reason why it should be administered as a continuous infusion by a pump via an indwelling central venous catheter. Epoprostenol was the first medication tested in a randomised trial in PAH and showed improvement of survival in these patients.32 Flushing, headache, jaw pain or diarrhoea are the most frequent adverse reactions. The most serious side effect is infection of the catheter and sepsis. Treatment with epoprostenol can be very challenging as it requires education of the patients and their carers on handling with infusion using aseptic technique. At the same time, stopping the infusion once started may precipitate rebound PH and death. On the other hand, epoprostenol IV infusion has no maximum dose being limited by side effects. Patients can increase the dose according to their symptoms providing it is well tolerated.
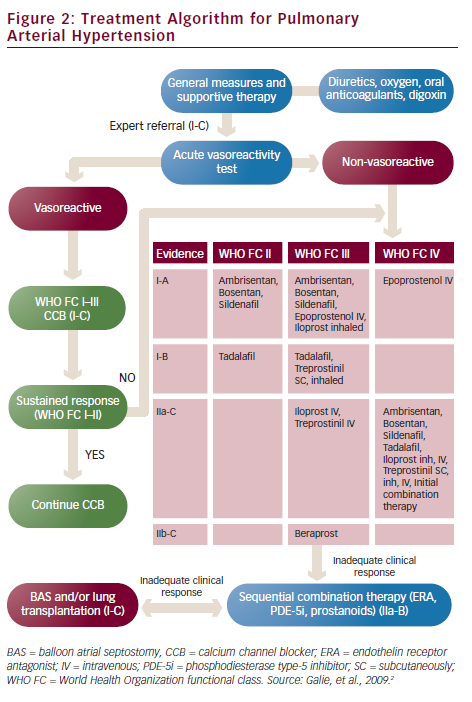
Iloprost is a chemically stable prostacyclin analogue that can be administered IV, orally or as an inhaler. Its major drawback when nebulised is the need of frequent administrations, as it should be given 6–9 times daily. Treprostinil can be given subcutaneously (SC), IV or as an inhaler. The SC administration of the drug is done by a pump via a small SC catheter and frequently is complicated with pain at the site of the infusion. When the patient fails to meet the targets of the treatment, then combination therapy with drugs of the three classes are recommended. Combination therapy has been shown to be beneficial in many clinical trials.30,33–35 A more aggressive initial treatment strategy with upfront dual combination therapy in selected PAH patients is currently being tested in the AMBITION (A study of First-Line Ambrisentan and Tadalafil Combination Therapy in Subjects With Pulmonary Arterial Hypertension [PAH]) trial.36 When all the treatments fail to improve patients’ symptoms and functional capacity then transplantation is the last therapeutic option. Double-lung is usually preferred over heart-lung transplantation because of the shortage of donor organs.
Calcium Channel Blockers
In the case of a positive vasoreactivity test, high doses of CCB should be preferred. Nifedipine, diltiazem and amlodipine are the agents most frequently used. Reassessment of a patient on CCB after treatment for 3–4 months with a RHC is necessary in order to clarify if there is a satisfactory response to CCB. True vasoreactivity usually leads to near-normalisation of pulmonary pressures.
Supportive Treatments
Apart from PAH-targeted treatments, supportive treatments such as oral anticoagulants, diuretics and oxygen are of importance. Evidence for oral anticoagulation in PAH patients is limited only to IPAH, heritable PAH and PAH associated with anorexigens, as small retrospective trials have shown. Nonetheless, the majority of PH experts tend to anticoagulate most of the PAH patients when the risk of bleeding is not increased. In PAH patients on IV epoprostenol, anticoagulation may also prevent catheter-associated thrombosis. A major issue related to PAH patients’ care is the high risk of pregnancy. As maternal mortality can reach up to 30–50 % during pregnancy, contraception is strongly recommended. Air travel or ascending to altitude more than 1,500–2,000 m can lead to severe hypoxia and supplemental oxygen is recommended, especially if the patient is in WHO FC III or has a PaO2<8 KPa.
New Treatments
New treatments are being developed and tested in ongoing trials. The new drugs either act on the established pathways already targeted by existing PAH-specific therapies or new agents target novel pathogenic pathways. Macitentan,37 an ERA, selexipag,38 an oral prostacyclin analogue, oral treprostinil,39 riociguat,40 an agent acting on the NO pathway, imatinib,41 a tyrosine kinase inhibitor and serotonin receptor antagonists42 are some of them.
Conclusions
PAH is a progressive and fatal disease, which may rapidly lead to right ventricular failure and death if left untreated. In the Registry to Evaluate Early And Long-term PAH Disease Management (REVEAL registry™) one in five patients with PAH reported symptoms >2 years before their disease was recognised.43 That delay reflects a similar delay for treatment and makes the early diagnosis of the disease absolutely crucial. General practitioners, cardiologists and respiratory physicians should always bear PAH in mind as a possible diagnosis when patients’ symptoms don’t improve with conventional therapies for breathlessness. Referring these patients to PH expert centres is also important as investigations can often be challenging and PAH-targeted treatments should be prescribed and monitored by PH specialists. On the other hand, PH specialists should promptly recognise patients’ clinical deterioration and always try to treat-to-target by moving from one stage of treatment to the other proactively. A high level of knowledge and clinical alert from all the physicians who are involved in managing patients with PH could result in a better prognosis by modifying the dismal natural history of the disease.