










Induced Pluripotent Stem Cells and Their Potential Applications
Induced pluripotent stem cells (iPSCs) are generated from somatic cells, such as skin fibroblasts, by ectopic expression of defined reprogramming factors. Within a few years of the first report of the generation of mouse iPSCs, several laboratories reportedly reproduced these cells using other cell types and species using similar approaches.1–4 This early attention on reproducible methods for the production of iPSCs from mammalian cells accelerated research into iPSC technology for clinical applications. iPSCs show unlimited proliferation capacity and pluripotency, as observed in embryonic stem cells (ESCs), and thus have significant advantages as a cell source for producing sufficient numbers of any cell type. In contrast with ESCs, human (h) iPSCs can be established from differentiated cells without destroying human embryos, thereby overcoming related ethical issues. Thus, iPSCs have been extensively investigated worldwide for applications in disease modelling, drug screening and regenerative medicine (Figure 1).2,5
When hiPSCs are derived from patients with a genetic disease caused by a mutation, such patient-derived iPSCs are called disease-specific hiPSCs. As disease-specific hiPSCs contain the same genetic information as the patient, including mutations corresponding to the altered gene function,6,7 disease-specific hiPSCs could potentially be a powerful tool for modelling human disease. Particularly in cardiovascular research, obtaining a sufficient number of cardiomyocytes (CMs) from patients is challenging due to the highly invasive procedures required to extract them. Further, the low proliferation capacity of CMs limits researchers’ ability to maintain these cells in culture. Being able to generate iPSC-derived CMs (hiPSC-CMs) from a specific patient overcomes this problem, and enables identification of typical cellular responses to pathological stress and therapeutic agents because these cells potentially reflect the biological responses of an individual patient’s own CMs (Figure 1).
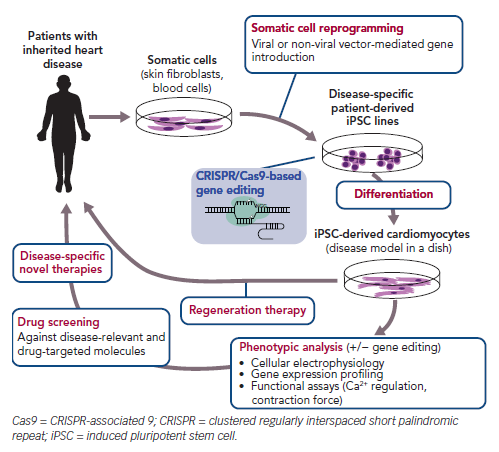
Recent genetic research has led to the identification of gene mutations responsible for hereditary heart diseases. Investigations into the pathophysiology of those inherited diseases often use animal models that partially mirror the disease conditions. However, animal studies are low throughput, time consuming and relatively expensive. Moreover, there are interspecies differences between humans and the experimental animals in terms of molecular and physiological properties (e.g. ion channel expression profile, heart rate), as well as in the cellular responses to pathological stress. Therefore, experimental results obtained from animal models do not perfectly recapitulate the conditions occurring in humans, and are less reliable for the purpose of extrapolation. In contrast, disease-specific hiPSCs could be a valuable tool in research on inherited diseases and for testing therapeutic agents. hiPSCs are created from somatic cells, which can be easily collected from accessible patient tissues, such as skin and blood. Owing to their self-renewal property, hiPSCs could be used to produce a sufficient number of specific cell types following appropriate differentiation methods for further experiments in vitro.
Human Induced Pluripotent Stem Cells for Modelling Inherited Arrhythmias
Advances in cardiovascular research have increased our understanding of the molecular mechanisms underlying various genetic diseases. Comprehensive genetic studies have identified causal mutations responsible for phenotypes of inherited cardiovascular diseases such as long QT syndrome (LQTS), Brugada syndrome and cardiomyopathies.
LQTS is characterised by a significantly prolonged QT interval attributable to delayed repolarisation in the ventricular myocardium. Some types of LQTS cause life-threatening arrhythmias in response to stimuli such as swimming and sudden loud noise. Genetic studies have found a number of gene loci responsible for LQTS in families with a high incidence of the disease. Despite an absence of clinical symptoms under sedentary conditions in patients with LQTS, once ventricular tachyarrhythmias are triggered by specific stimuli, patients with LQTS are prone to exhibit syncope. Sustained arrhythmias ultimately lead to VF, resulting in sudden cardiac death. Several studies on patients with LQTS have identified a number of mutations in genes encoding cardiac ion channels, which are membrane proteins regulating the generation and propagation of action potential.8–10 However, these mutations are not always responsible for the observed symptoms, even when the patients are exposed to the stimuli that trigger electrophysiological changes.
Effects of the stimuli or therapeutic agents, as well as the incidence of cardiac events, vary considerably among individual patients. Therefore, to address issues related to proarrhythmic mechanisms in individuals with inherited LQTS, patient-derived hiPSC-CMs with the corresponding mutation(s) could serve as powerful tools for in vitro experiments. Previous studies characterising mutations of the alpha-subunit of the potassium voltage-gated channel subfamily Q member 1 (KCNQ1; also known as KVLQT1 and KV7.1) using patient-derived iPSC-CMs revealed that impaired membrane trafficking of Ks channels and reduced delayed rectifier potassium channel current (IKr) cause LQT1.9,11,12 Itzhaki et al. introduced reprogramming factors into dermal fibroblasts obtained from patients with a mutation in the alpha-subunit of potassium voltage-gated channel subfamily H member 2 (KCNH2; responsible for IKr) causing LQT2.13 Spontaneously beating hiPSC-CMs carrying this mutation were used for functional analysis and exhibited a prolonged QT interval similar to that in LQTS patients.
Similar studies using hiPSC-CMs derived from a patient with a missense mutation in KCNH2 also exhibited action potential prolongation, smaller IKr, early afterdepolarisations and arrhythmias. These changes were recovered or exaggerated by pharmacological agents or selective RNA interference in disease-specific hiPSC-CMs.13–16
Disease-specific hiPSC-CMs from patients and families with Timothy syndrome (LQT8) that have a mutation located in calcium voltage-gated channel subunit alpha1 C (CACNA1C; responsible for the L-type calcium current, ICa,L) have been established and assessed for mutation-associated phenotypes in vitro.10,17,18 An LQT8 model using patient-specific hiPSC-CMs reflected cellular electrical abnormalities, including prolonged action potential duration, delayed afterdepolarisations and altered Ca2+ transients. In contrast, roscovitine, an inhibitor of cyclin-dependent kinase 5, a key mediator involved in the regulation of CaV1.2 channels, enhanced ICa,L inactivation, shortened action potential duration, restored the irregular Ca2+ transient and decreased the frequency of abnormal depolarisations in LQT8 hiPSC-CMs.10,17,18
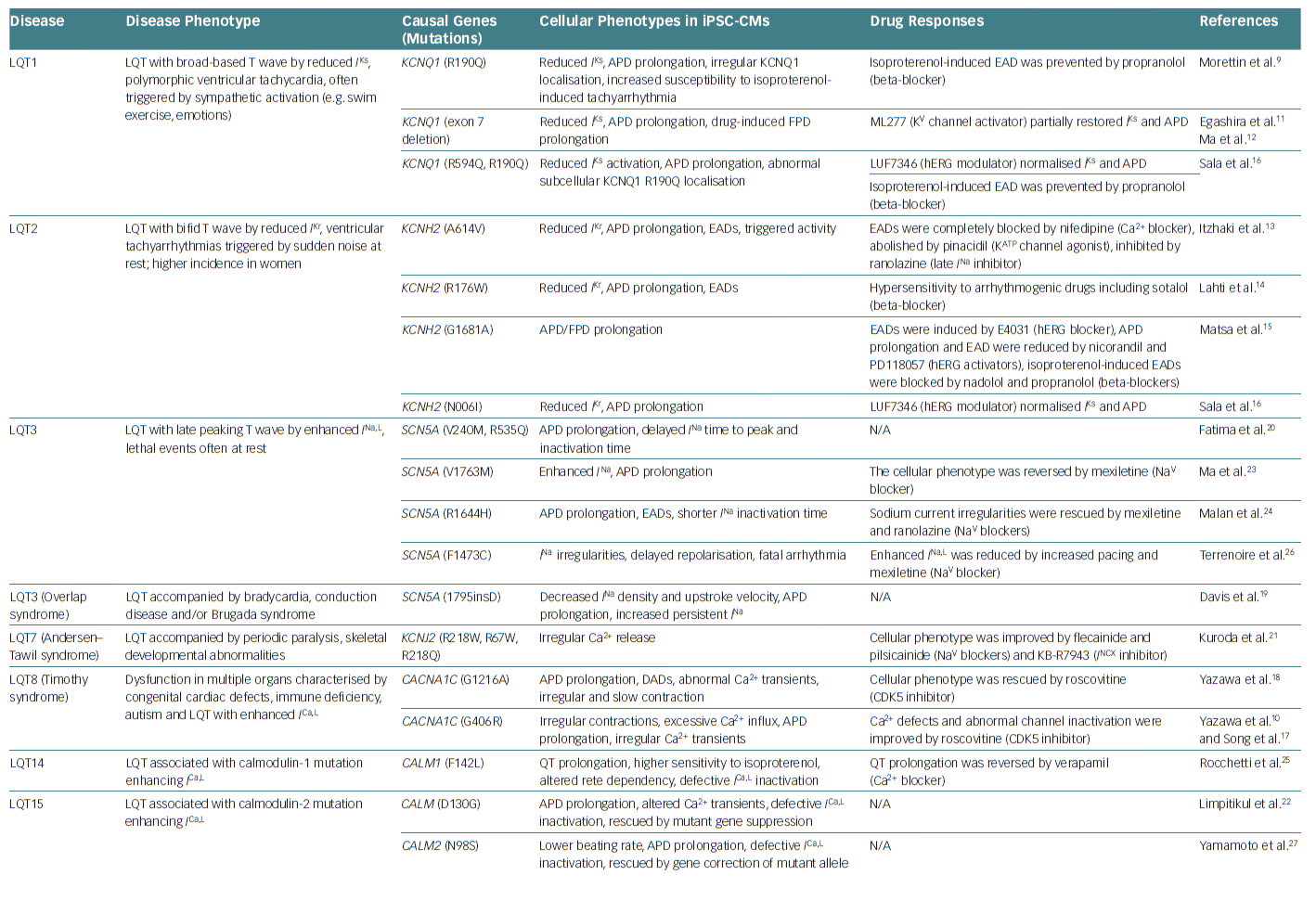
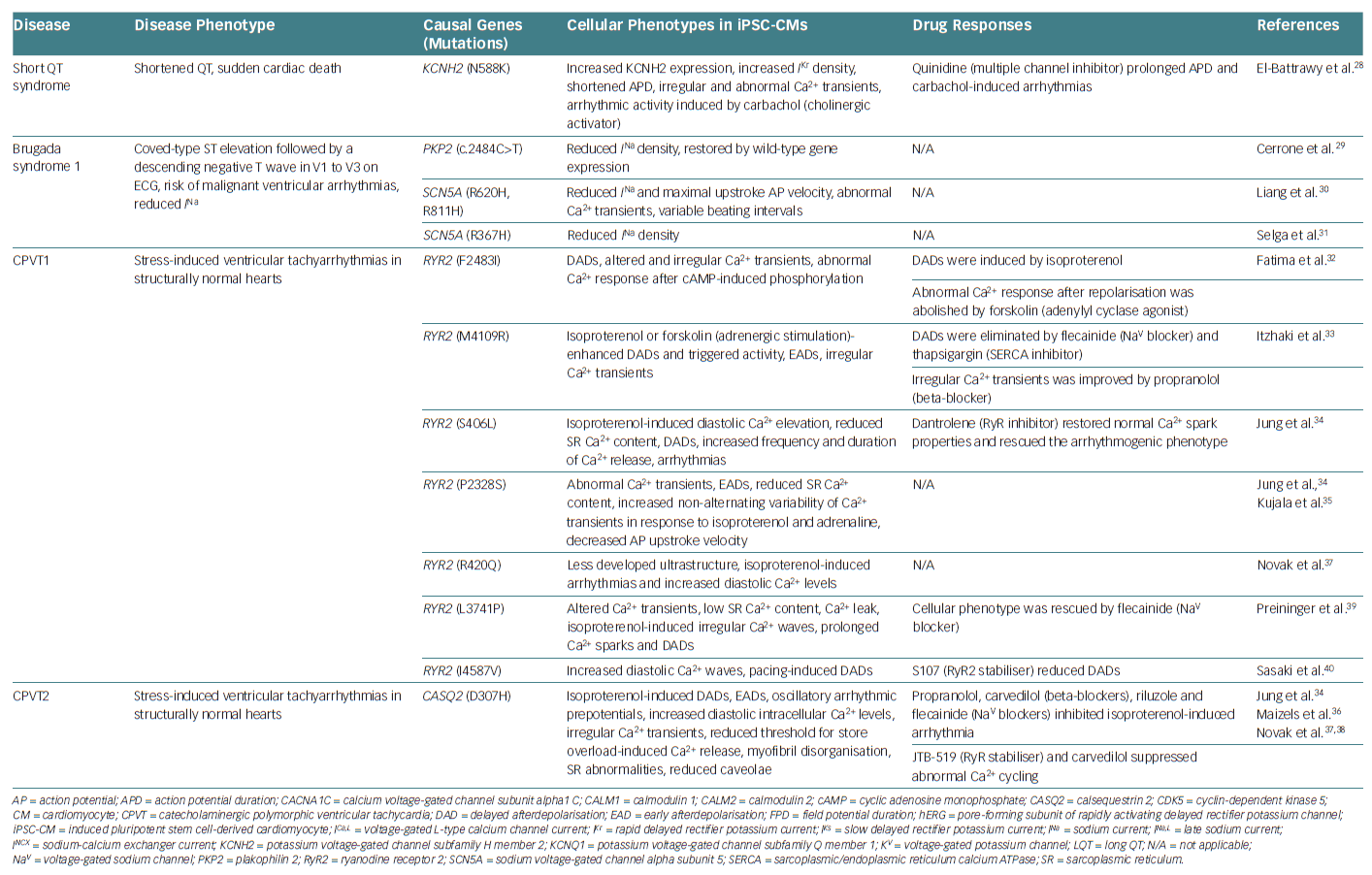
Furthermore, other inherited arrhythmias have been investigated using disease-specific hiPSC-CMs, including various types of LQTS – mutations in sodium voltage-gated channel alpha subunit 5 (SCN5A), potassium inwardly rectifying channel subfamily J member 2 (KCNJ2), calmodulin 1 (CALM1) or calmodulin 2 (CALM2), short QT syndrome (KCNH2 mutation), Brugada syndrome type 1 (SCN5A mutation) and catecholaminergic polymorphic ventricular tachycardia (mutations in ryanodine receptor 2 (RYR2) or calsequestrin 2 [CASQ2]).19–40 These cells recapitulated cellular electrophysiological changes in the heart of patients. Table 1 summarises the different studies that have used hiPSC-CMs as models to investigate inherited arrhythmias.
Human Induced Pluripotent Stem Cells for Modelling of Inherited Cardiomyopathies
In addition to inherited arrhythmias, there are some incidences of cardiomyopathies in families carrying specific genetic variant(s) that are responsible for causing the disease. Dilated cardiomyopathy (DCM) is a major type of cardiomyopathy that is characterised by systolic dysfunction and dilated cardiac chambers comprised of thin myocardial walls.41 Most cases of DCM without any identifiable cause (e.g. coronary artery disease, systemic hypertension, viral infection) are diagnosed as ‘idiopathic’ DCM.
Based on family history and clinical findings, including sudden cardiac death, heart failure and abnormal echocardiography, previous clinical studies have proposed that familial transmission of idiopathic DCM is observed in 20–50% of patients.42–44 When idiopathic DCM is identified in two or more family members, it is defined as familial DCM (FDC). FDC is largely caused by autosomal dominant mutations in key cardiac genes encoding sarcomere-related proteins, cytoskeletal proteins, mitochondrial proteins, nuclear membrane proteins and calcium regulators.43,45,46 These loss-of-function mutations lead to the abnormal morphology and function of the heart that is seen in idiopathic DCM. Moreover, recently developed high-throughput gene analyses have revealed that inherited DCM is associated with mutations in more than 100 gene loci.47
Although the pathophysiology of FDC is heterogeneous, the effect of each individual mutation has been unclear in the context of FDC. To address this, human CMs are ideal for in vitro functional analysis of mutations associated with FDC, but, as mentioned earlier, it is difficult to acquire a renewable source of cardiac cells. Compared with animal models and non-CMs expressing DCM mutant proteins, hiPSC-CMs are expected to exhibit responses similar to those observed in native human myocardium. For example, individual families carry a mutation that causes an arginine-to-tryptophan substitution at amino acid position 173 in the cardiac troponin T (cTnT) protein.48 Patient-specific hiPSCs were produced using minimally invasive procedures from skin fibroblasts of family members, and hiPSC-CMs were generated and tested to investigate the mechanisms underlying FDC. The FDC hiPSC-CMs exhibited reduced Ca2+ influx and contractility, despite normal electrophysiological properties. These cells also showed the characteristic patchy structure of myofilaments, which was enhanced upon noradrenaline stimulation and stretching, leading to systolic dysfunction.48
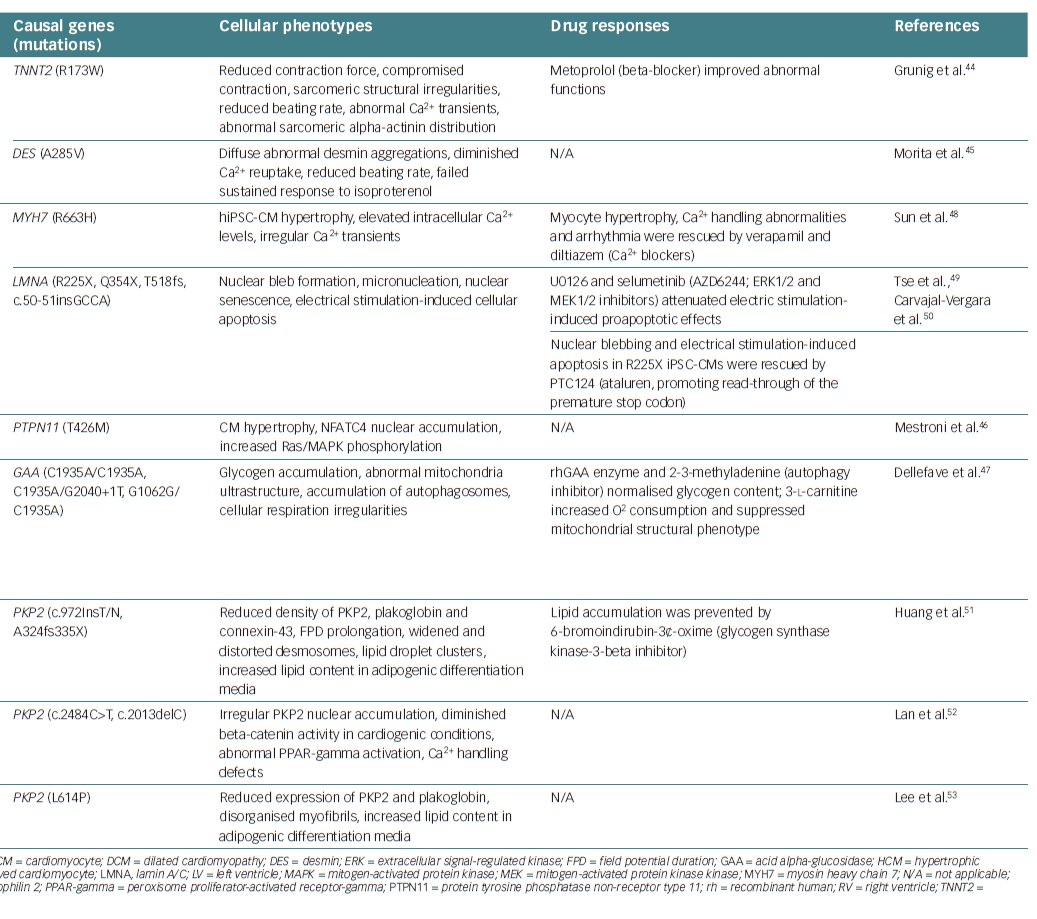
This is consistent with the fact that the tendency towards DCM is enhanced by increases in inotropic effects and hypertension. These findings explain the involvement of cTnT dysfunction in the development of DCM. Thus, FDC hiPSC-CMs recreate, at least in part, the pathophysiology of FDC in human patients. Other causal gene mutations responsible for inherited cardiomyopathies, including DCM, hypertrophic cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy/dysplasia, have been reported.49–57 Table 2 lists studies that have used hiPSC-CMs as models for investigating inherited cardiomyopathies.
Although numerous studies have summarised the characteristic features of familial heart diseases using patient-specific hiPSC-CMs, as described above, it is still challenging to fully recapitulate the disease phenotype using iPSC-CM-based disease modelling, primarily because hiPSC-CMs exhibit immature functions and morphology. For example, an incomplete ion channel profile (e.g. lack of IK1, corresponding to slower action potential kinetics and a relatively positive diastolic potential) and subcellular structure (e.g. the absence of or underdeveloped T-tubule and sarcomere formation) are commonly observed in hiPSC-CMs.58–60 The gene expression profile of hiPSC-CMs also resembles that of foetal CMs and is distinct from that of adult CMs.60,61 The immaturity of hiPSC-CMs in terms of function and gene expression profile may result in controversial findings, particularly in the investigation of late-onset cardiac diseases that largely require adult CM-like cells for disease modelling.
In an in vitro study using hiPSC-CMs to investigate the pathophysiology of late-onset Pompe disease, which is characterised by slow progression of muscle weakness, although patient-specific hiPSC-CMs exhibited typical features associated with the disease, such as intracellular glycogen accumulation and mitochondrial dysfunction, they did not fully exhibit the autophagic abnormalities that are observed in vivo.62,63 This may be overcome by using fully differentiated hiPSC-CMs assembled along with a complete subcellular system for muscle contraction, Ca2+ cycling, metabolism and protein recycling. Recent studies have contributed to the development of protocols for the maturation of hiPSC-CMs using electrical and/or mechanical stimulation, a 3D culture system with scaffold materials, coculture with fibroblasts or CMs in vitro and in vivo and a combination of these techniques, leading to improvement in contractility, Ca2+ handling and electrophysiological properties.64–68
Lack of chamber-specific characteristics is another major concern regarding the use of hiPSC-CMs for disease modelling. As the structure, haemodynamic stress, developmental origin and protein expression profile are quite distinctive among the cardiac chambers,59,69,70 the molecular features of individual CMs in each chamber would also differ. Some inherited arrhythmias and cardiomyopathies have chamber-specific characteristics. Clinical phenotypes of Brugada syndrome and ARVC/D likely originate from the right ventricular outflow tract. However, disease models based on hiPSC-CMs may not fully recapitulate the characteristic features of any specific region of the heart.
A differentiated hiPSC-CM cluster usually consists of electrophysiologically heterogeneous subtypes including ventricular-, atrial- and nodal-like myocytes. The ventricular-like hiPSC-CMs exhibit properties analogous to those of human ventricular myocytes (e.g. steep upstroke (Phase 0) and plateau phase (Phase 4) of action potentials), whereas the nodal-type hiPSC-CMs exhibit slower action potential kinetics and depolarising diastolic potential.71 This mixed subtype of hiPSC-CMs leads to a wide range of results rather than being representative of a specific subtype of CMs. The development of protocols for subtype-specific and/or chamber-specific differentiation of hiPSC-CMs will accelerate research to identify the chamber-specific phenotypes associated with heart diseases. Although some genetic heart diseases are rare, many of them lead to life-threatening conditions. Therefore, further intensive research using disease-specific hiPSC-CMs should be promoted to gain insights into the underlying mechanisms and to identify potential therapeutic targets of these genetic diseases in order to develop novel therapeutic approaches for individual patients.
Human Induced Pluripotent Stem Cells as a Tool for Drug Screening
Currently, the development of new drugs requires multiple processes, including screening of numerous putative drug compounds based on chemical structure and in vitro assays of pharmacological activity, followed by analyses of pharmacokinetics and safety in vitro and in vivo and, finally, clinical trials in humans. In most cases, these processes take many years until the candidate compounds are tested in humans.72 Even though the effectiveness of compounds may be promising in cell culture and animal experiments, problems identified in clinical trials assessing the effects of these compounds on the QT interval (known as a thorough QT/QTc study) following pharmacokinetics examination in humans may halt the further development of these compounds. However, if human cardiac cells were widely available, drug testing in human CMs might provide effective and safe drug candidates rapidly and economically, because the response to compounds tested using in vitro experiments with human CMs could resemble that of the human body.
Disease-specific hiPSC-derived CMs potentially exhibit similar physiological characteristics as diseased cells in patients, and may be a useful tool to predict the benefits and side-effects of drug candidates in patients. Drug screening using hiPSC-CMs to detect side effects such as drug-induced QT prolongation and ventricular tachyarrhythmias could contribute to the early withdrawal of therapeutic compounds with undesirable cardiac effects before the initiation of in vivo experiments and clinical trials.72,73 Other than the development of new drugs, the cardiac side effects of some already marketed drugs, including anti-arrhythmic drugs and non-cardiac drugs such as antihistamines, antipsychotics and anti-infective drugs, have been widely recognised. These drugs have the potential to cause torsade de pointes, in combination with other endogenous and environmental factors.73 Drug testing using hiPSC-CMs may also be applicable in this context.
Although hiPSC-CMs share some characteristics with adult human ventricular myocytes, hiPSC-CMs are commonly known to exhibit the features of foetal ‘immature’ CMs in terms of their gene expression profile, structure and electrophysiology, as noted above. hiPSC-CMs express cardiac-specific genes (e.g. those encoding cTnT, alpha-myosin heavy chain) and exhibit ion channel activity (e.g. similar INa, IKr and ICa,L current density to that in adult ventricular CMs);12–14,16,71,74–83 however, morphologically they are more rounded or multiangular in shape and smaller in size, with disorganised myofibrils and a lack of t-tubules, which contribute to the slower kinetics of the Ca2+ transient.38,76,83–87 These important differences should be considered when using hiPSC-CMs in drug screening. Further investigations are needed to develop optimal methods for more efficient differentiation into functional CMs that exhibit the typical properties of adult CMs.
Gene Editing to Create Disease-Specific Human Induced Pluripotent Stem Cells
Comprehensive genetic studies have identified causal mutations responsible for genetic heart diseases. hiPSC-CMs have emerged as a highly effective tool for modelling such diseases. Although it is technically possible to induce disease-specific hiPSC-CMs, patient-derived somatic cells may not be readily available, especially in the case of rare diseases. In addition, interclonal variation is seen among hiPSC clones, resulting from different genetic backgrounds associated with individual cells.
Clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein (Cas) 9 is a gene-editing technology that can solve the challenges associated with the genetic variability.88,89 CRISPR is a DNA sequence found in bacterial genomes; it is thought to be derived from viruses, is known to protect bacteria from repeated viral infections and acts as a basic adaptive immune system for prokaryotes. Cas9 is a DNA-cutting enzyme that recognises CRISPR sequences and causes site-specific DNA double-strand breaks (Figure 2). Recent advances in CRISPR/Cas9-based gene editing have markedly improved the efficiency and specificity of the method and expanded its applications, including knockout, repression and activation of genes of interest.90
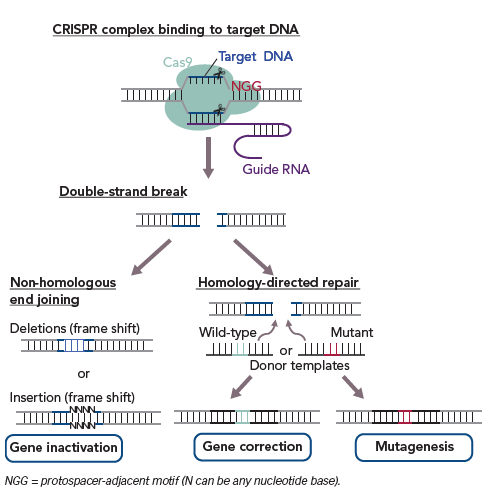
In phenotypic analysis of monogenic inherited diseases, this technology is also applicable to either disease-associated mutagenesis in wild-type hiPSCs or to the correction of pathogenic gene mutations in disease-specific hiPSCs (Figure 3).89 Analysis of disease-specific hiPSCs versus wild-type hiPSCs established from healthy donor cells as a control may result in unreliable outcomes due to the different genetic backgrounds of the disease-specific hiPSCs and control cells. However, CRISPR/Cas9-based gene editing enables the preparation of an isogenic control by normalising a disease-relevant mutation in disease-specific hiPSCs or by inducing the mutation in wild-type hiPSCs so that diseased and control cells with the same genetic background are obtained. In addition, CRISPR/Cas9-based gene editing could allow the production of isogenic cells with intact and/or corrected variant alleles in non-coding regions including enhancers that may reveal the role of mutations in the transcriptional regulation of genes responsible for a disease phenotype. This method shows promise for the proper evaluation of the involvement of mutated genes in disease phenotype following in vitro differentiation (Figure 3).
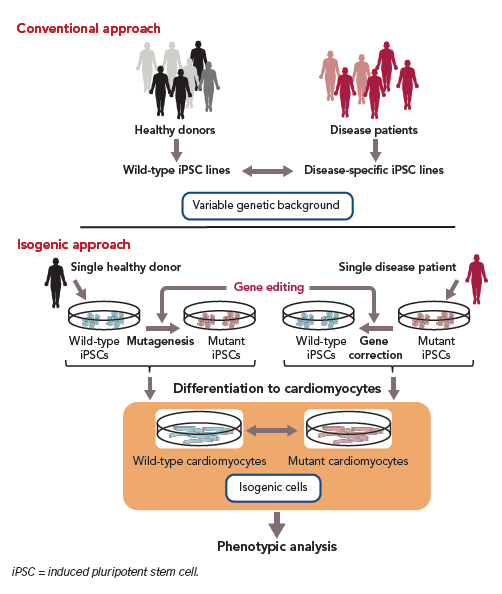
Polygenic diseases, which differ from monogenic inherited diseases in that more than one gene is involved in their dysfunction, impose another limitation on the use of hiPSCs. Polygenic diseases are thought to be caused by a combination of multiple mutations, each of which has a small effect, with or without extrinsic factors. Although gene editing has been used to edit multiple regions of the genome, a major challenge towards using hiPSCs to investigate polygenic diseases is identification of the corresponding mutations and understanding how each mutation contributes to the pathogenesis of these multifactorial diseases. Moreover, in some cases, environmental factors may strongly affect disease phenotypes, making experimental conditions and further analysis more complicated. Comprehensive reviews are available for detailed information regarding the use of gene editing in iPSC research.89,91
Consideration of Human Induced Pluripotent Stem Cells for Application in Disease Modelling and Clinical Use
Despite extensive benefits, there are still many unsolved issues regarding the use of hiPSCs in further applications. One of the major issues is that the quality of individual hiPSC lines is variable, even when an hiPSC line is derived from one individual. Classical iPSC reprogramming methods using retroviral or lentiviral vectors may cause random insertional mutations in the host genome, resulting in alteration of subsequent cell phenotypes.92
Recent advances in reprogramming strategies using non-integrating, virus-free and vector-free methods are overcoming this issue.93,94 However, it is still technically difficult to eliminate the risk of gene mutations during the reprogramming process because forced expression of reprogramming factors can induce DNA damage.95 In fact, protein-coding point mutations acquired during or after reprogramming were identified in multiple hiPSC lines, some of which exhibit unpredictable phenotypes.96 Thus, accumulating evidence regarding the mechanism underlying the reprogramming of iPSCs is expected to provide insights into how the quality of hiPSC lines may be stabilised and standardised for use as a cell source for further experiments and clinical application.
Precise investigations into the pathophysiology of inherited diseases using patient-derived iPSCs require improved protocols that allow highly efficient differentiation of hiPSCs into a specific cell type, because the differentiation efficiency in current experiments remains significantly lower than what is desired. The characteristic variability of cells differentiated from disease-specific hiPSCs is a considerable hurdle that research into pathophysiology must overcome. Epigenetic modifications are presumably one of the causes of phenotype variability. Optimised sorting methods to collect only a desired cell type from the heterogeneous cell population need to be developed. Current research efforts are advancing cardiac differentiation protocols to generate spontaneously beating CM-like cell clusters, but the clusters of differentiated cells that are heterogeneous also contain other mesodermal derivatives, such as smooth muscle cells and endothelial cells, as well as undifferentiated cells, which may increase the risk of tumourigenesis.
Pathophysiological studies using disease-specific hiPSCs allow us to determine the cellular characteristics of a disease, but do not recreate the function of the whole organ within the body. Although complex bioengineering approaches, such as organoid formation and 3D culture systems, are available,97,98 it is difficult to use these methods in the heart because CMs in the heart are predominantly situated in a highly organised structure comprising vessels, nerves, mesenchymal cells, extracellular matrix and myocytes. In addition, CMs are continuously exposed to dynamically changing neuroendocrine factors and mechanical stresses. Therefore, it should be considered that studies using disease-specific hiPSC-CMs fundamentally provide simplified information regarding the pathophysiology in patients with a familial disease. Nevertheless, the experimental data from these cells may reveal responses that mirror actual phenomena in human patients, and are thus valuable for gaining an understanding of the inherited disease.
Conclusion
Disease-specific hiPSC-CMs, which carry the same genomic information as patients with inherited diseases, can undoubtedly be of use in research to address the pathophysiology of monogenic inherited diseases, the drug responsiveness of patients for personalised medicine and drug development by providing a cell source for screening compounds and drug safety testing. A combination of disease-specific hiPSC-CMs and gene-editing technologies may further advance our understanding of genetic diseases and drug development in cardiovascular medicine.