Purpose: Tyrosine kinase inhibitors (TKI) are known to induce cardiac toxicity in patients, which may be caused by mitochondrial damage. The aim of the current study was to improve our knowledge about the role of mitochondria in cardiac toxicity of TKIs, in particular concerning oxidative stress and cell death.
Design and methods: We exposed cardiac H9c2 cells for 24 hours with imatinib, sorafenib or sunitinib. In addition, we treated mice with sunitinib (7.5 mg/kg/day) for 2 weeks.
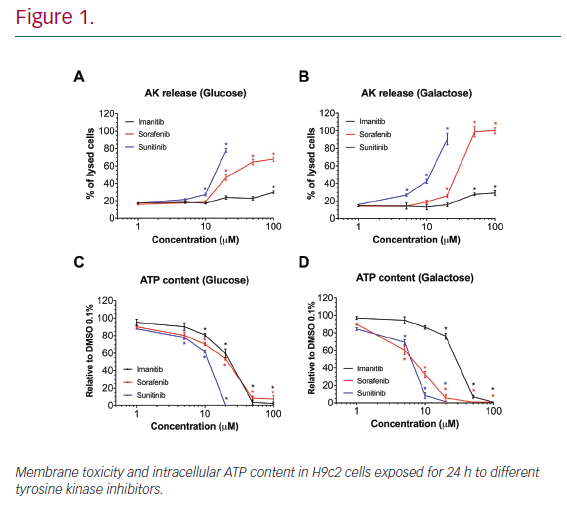
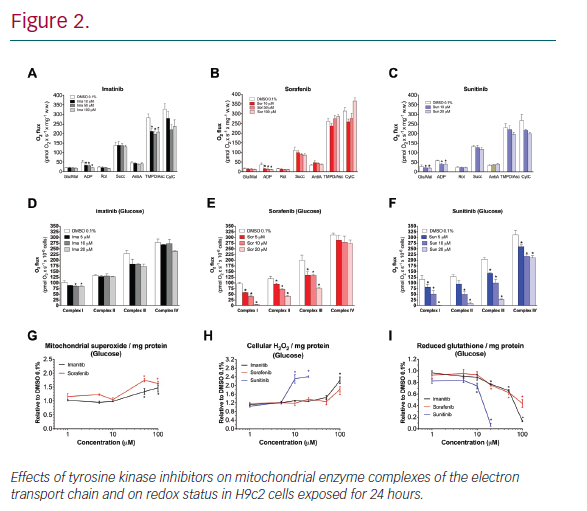
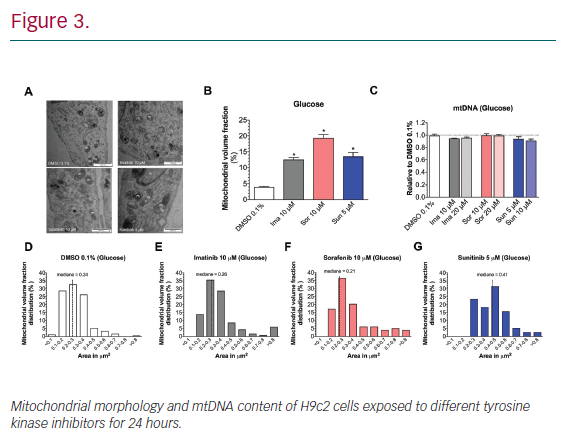
Results: In H9c2 cells exposed for 24 hours, all TKIs investigated showed a higher cytotoxicity profile in the presence of galactose (favouring mitochondrial metabolism) compared to glucose (favouring glycolysis). The TKIs dissipated the mitochondrial membrane potential and reduced activities of mitochondrial enzyme complexes of the electron transport chain (ETC). In addition, the TKIs increased superoxide accumulation and decreased the cellular GSH pool, inducing apoptosis. Electron microscopy showed swollen mitochondria with loss of cristae. In mice, treatment with sunitinib for 2 weeks increased plasma troponin I and creatine kinase MB, indicating cardiomyocyte damage. The activity of enzyme complexes of the ETC was decreased and the mitochondrial content of reactive oxygen species (ROS) was increased. Cleavage of caspase 3 was increased in hearts of sunitinib-treated mice, suggesting cardiomyocyte apoptosis.
Conclusion: Mitochondrial ROS accumulation appears to be an important mechanism of cardiotoxicity associated with sunitinib and other cardiotoxic TKIs, leading to the activation of apoptosis.